Abevmy
Brand Information
| Brand name | Abevmy |
| Active ingredient | Bevacizumab, Bevacizumab |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Abevmy.
Summary CMI
ABEVMY®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I receiving ABEVMY?
ABEVMY contains the active ingredient bevacizumab (rch). ABEVMY is used to treat some cancers in the brain, lung, bowel, kidney, breast, or female reproductive system. For more information, see Section 1. Why am I receiving ABEVMY? in the full CMI.
2. What should I know before I am given ABEVMY?
Read this leaflet carefully before you use ABEVMY and keep it with the medicine. Check the list of ingredients at the end of the CMI. Do not use ABEVMY if you have ever had an allergic reaction to any of the ingredients.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I am given ABEVMY? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with ABEVMY and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How will I be given ABEVMY?
You will be given ABEVMY as an infusion (slow injection or "drip") into a vein by a health care professional every 2 to 3 weeks. The number of infusions you will receive depends on how you are responding to treatment. Your doctor will discuss this with you.
The infusion usually takes between 30-90 minutes.
More instructions can be found in Section 4. How will I be given ABEVMY? in the full CMI.
5. What should I know while being treated with ABEVMY?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
For more information, see Section 5. What should I know while being treated with ABEVMY? in the full CMI.
6. Are there any side effects?
Common side effects include weakness, pain, headache, bleeding, nose bleeds, nausea, vomiting, diarrhoea, loss of appetite, and mouth sores, rectal bleeding, bleeding or bruising more easily than normal, fainting. Serious side effects include serious bleeding, severe high blood pressure, holes in the stomach or intestine, A stroke, heart problems, nervous system or vision problems, shortness of breath, difficulty breathing, or fistula (abnormal connection between two body parts). For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
ABEVMY®
Active ingredient(s): bevacizumab (rch)
Consumer Medicine Information (CMI)
This leaflet provides important information about using. ABEVMY. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using ABEVMY.
Where to find information in this leaflet:
1. Why am I receiving ABEVMY?
2. What should I know before I am given ABEVMY?
3. What if I am taking other medicines?
4. How will I be given ABEVMY?
5. What should I know while being treated with ABEVMY?
6. Are there any side effects?
7. Product details
1. Why am I receiving ABEVMY?
ABEVMY contains the active ingredient bevacizumab.
ABEVMY is a biosimilar medicine to AVASTIN®. It has been assessed to be as safe and effective as the reference product and provides the same health outcomes. Avastin® is not available in Australia.
ABEVMY is used to treat:
- Brain tumours that are resistant to previous treatments.
- Metastatic cancers of the large bowel (i.e., in the colon or rectum), breast or cervix in combination with chemotherapy agents. Metastatic means the cancer has spread to other parts of the body from where it started.
- Lung cancer and cancer of the ovaries and fallopian tubes (which can extend to the lining of surrounding organs such as stomach, liver) in combination with chemotherapy agents.
- Kidney cancer in combination with interferon therapy (Roferon-A®).
All cancers need a blood supply to be able to survive and grow. Bevacizumab targets a protein called vascular endothelial growth factor (VEGF). This protein helps cancer cells to grow blood vessels, so they can get nutrients and oxygen from the blood. When ABEVMY binds to VEGF it disrupts the blood supply to the tumour, stopping or slowing down its growth.
2. What should I know before I am given ABEVMY?
Warnings
ABEVMY is not approved for use in the eye.
- Based on your age, especially if you are 65 years or older, or on your general health, your doctor will discuss your ability to tolerate ABEVMY.
- ABEVMY can increase the risk of blood clots which can lead to strokes or heart attacks in patients older than 65 years of age compared with younger patients.
- ABEVMY can also increase the risk of side effects in patients older than 65 years of age compared with younger patients.
You must not be given ABEVMY if you:
- are allergic to bevacizumab, or any of the ingredients listed at the end of this leaflet. Always check the ingredients to make sure you can use it. Symptoms of an allergic reaction may include shortness of breath; wheezing or difficulty breathing; swelling of the face, lips, tongue or other parts of the body or rash, itching or hives on the skin.
- have had an allergic reaction to any proteins that are of Chinese hamster origin or to other recombinant human or humanised antibodies.
- are a child or adolescent. The safety and effectiveness in children and adolescents have not been established.
Check with your doctor if you:
- have or previously have had any of the following:
- radiation therapy or radiotherapy (treatment with high energy x-rays)
- inflammation of the bowel (symptoms may include fever, vomiting, diarrhoea and stomach pain) or stomach ulcers
- hypertension (high blood pressure) - it is important to follow all your doctor's instructions to control your blood pressure
- history of blood clots or stroke, or if you are taking medicine to prevent blood clots (e.g. warfarin) or if anyone in your family suffers from bleeding problems
- heart disease
- history of diabetes
- major surgery within the last 28 days or have a wound that has not healed properly. ABEVMY can cause an increased risk of post-operative bleeding or problems with wound healing.
- blocked lung artery (pulmonary embolism).
ABEVMY may increase the risk of recurrence
- received anthracyclines (e.g., doxorubicin) or have had radiotherapy to your chest. Anthracyclines are a specific type of chemotherapy used to treat some cancers. ABEVMY can increase the risk of developing a weak heart.
- pain in the mouth, teeth and/or jaw, swelling or sores inside the mouth, numbness or a feeling of heaviness in the jaw, or loosening of a tooth. Tell your doctor immediately. You may be advised to have a dental check-up before you start treatment with ABEVMY
- allergies to any other medicines, foods, dyes or preservatives
- other health problems.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy
Do not use ABEVMY if you are pregnant. ABEVMY may cause damage to your unborn baby.
Check with your doctor if you are pregnant or intend to become pregnant. ABEVMY may interfere with your ability to become pregnant. Your doctor will advise you of your options prior to starting treatment.
If you are a woman of childbearing potential, use suitable methods of contraception during treatment with ABEVMY and for at least 6 months after your last dose. Your doctor can advise about using contraception during treatment with ABEVMY.
Immediately inform your doctor if you become pregnant while you are being treated with ABEVMY.
Breastfeeding
Do not breastfeed while being treated with ABEVMY and for at least 6 months after the last dose. ABEVMY may interfere with the growth and development of your baby. Talk to your doctor if you are breastfeeding or intend to breastfeed.
3. What if I am taking other medicines?
Some medicines may interfere with ABEVMY and affect how it works.
Tell your healthcare professional if you:
- have recently received, or are receiving a bisphosphonate (medicine used in preventing or treating bones that are weak and likely to break), such as ibandronate sodium, zoledronic acid or disodium pamidronate.
- are taking other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Read the CMI leaflets of all medicines you take in combination with ABEVMY. This will help you understand the information related to those medicines.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect ABEVMY.
4. How will I be given ABEVMY?
When you will be given ABEVMY
ABEVMY will be given either once every 2 weeks or once every 3 weeks depending on the type of cancer to be treated.
How much ABEVMY you will be given
Your doctor will prescribe your dose. The dose depends on your body weight, general health and the type of cancer to be treated.
How ABEVMY is given
Your ABEVMY is given by infusion into a vein ("drip") by a health care professional. The first infusion is usually given over 90 minutes. If it is well tolerated the second infusion may be given over 60 minutes. Later infusions may be given over 30 minutes.
How long ABEVMY is given
The number of infusions you will receive depends on how you are responding to treatment. Your doctor will discuss this with you.
If you miss a dose
Your doctor will decide when you should be given your next dose of ABEVMY.
If you have been given too much ABEVMY
As ABEVMY is given to you under the supervision of your doctor, it is unlikely that you will receive too much.
However, if you experience severe side effects, like a severe migraine, after being given this medicine, tell your doctor or nurse immediately. You may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
5. What should I know while being treated with ABEVMY?
Things you should do
Be sure to keep all of your appointments with your doctor so that your progress can be checked.
Call your doctor straight away if you:
- become pregnant during treatment with ABEVMY, or
- plan to start a family in the near future.
- are breastfeeding while being treated with ABEVMY.
- are planning to have surgery.
- have a wound that is not healing properly.
- need to undergo an invasive dental treatment or dental surgery, in particular when you are also receiving or have received a bisphosphonate (for example medicines containing ibandronate sodium, zoledronic acid or disodium pamidronate)
- feel ABEVMY is not helping your condition.
Remind any doctor, dentist or pharmacist you visit that you are using ABEVMY.
Things you should not do
- Do not take any other medicines whether they require a prescription or not without first telling your doctor or consulting a pharmacist.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how ABEVMY affects you.
ABEVMY has not been shown to impair your ability to drive or operate machinery.
Looking after your medicine
ABEVMY will be stored in the pharmacy or on the hospital ward in a refrigerator at a temperature between 2-8°C.
Disposal
ABEVMY is for single use only.
The vials should be used once only, and any remaining contents should be discarded.
ABEVMY must not be used after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
Because ABEVMY is used with other medicines that treat cancer (including chemotherapy), it may be difficult for your doctor to tell whether the side effects are due to ABEVMY or due to other medicines.
ABEMVY may exacerbate some chemotherapy side effects when used in combination with chemotherapy agents including hair loss, nail disorders, pain, redness and/or swelling of your hands and/or soles of your feet, and a feeling of numbness or tingling in the hands or feet.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
Lungs and upper airways
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Infections and infestations
| Tell your doctor if you experience any of these side effects. |
Serious side effects
| Serious side effects | What to do |
Lungs and upper airways
| Tell your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Some side effects are more common in elderly patients. These include blood clots in the arteries, which can lead to a stroke or a heart attack. In addition, elderly patients have a higher risk of a reduction in the number of white cells in the blood and cells that help the blood clot, which can lead to infections and bleeding or bruising more easily than normal. Other side effects reported with a higher frequency in elderly patients were diarrhoea, nausea or sickness, headache, hair loss, inflammation of the mouth and throat, a feeling of numbness or tingling in the hands or feet and fatigue.
There have been reports of abnormal tube-like connections (fistulae) between internal organs and skin or other tissues that are not normally connected. You may have an increased risk of fistulae forming between the vagina and any part of the gastro-intestinal system if you are being treated with ABEVMY for cancer of the cervix.
ABVEMY is not approved to use in the eye. If injected directly into the eye additional side effects may occur including: infection (some cases leading to blindness), eye pain, redness of the eye, small particles or spots in your vision (floaters), seeing bright flashes of light with floaters, progressing to loss of sight, bleeding in the eye, cataracts, leading to surgery of the eye lens, serious side effects affecting other organs which can be severe or life-threatening and lead to hospitalization, e.g. stoke.
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What ABEVMY contains
| Active ingredient (main ingredient) | bevacizumab |
| Other ingredients (inactive ingredients) |
|
Do not take this medicine if you are allergic to any of these ingredients.
What ABEVMY looks like
ABEVMY is a clear to slightly opaque, colourless to pale brown solution.
ABEVMY is available as 100 mg and 400 mg single-dose vials.
ABEVMY bevacizumab 100 mg/4 mL concentrate injection for solution vial (AUST R 334816)
ABEVMY bevacizumab 400 mg/16 mL concentrate injection for solution vial (AUST R 334814)
Who distributes ABEVMY
Maxx Pharma Pty Ltd
Level 20, 181 William St,
Melbourne Victoria 3000
Phone: 1800 202 981
Email: medical@maxxpharma.com.au
This leaflet was prepared in March 2025.
Brand Information
| Brand name | Abevmy |
| Active ingredient | Bevacizumab, Bevacizumab |
| Schedule | S4 |
MIMS Revision Date: 01 May 2025
1 Name of Medicine
Bevacizumab (rch).
Abevmy is a biosimilar medicine to Avastin. The evidence for comparability supports the use of Abevmy for the listed indications.
Avastin is not available in Australia.
2 Qualitative and Quantitative Composition
Bevacizumab is the active ingredient.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Abevmy (bevacizumab) is available in 100 mg and 400 mg single dose vials containing 4 mL and 16 mL, respectively, of bevacizumab (25 mg/mL).
Abevmy is a clear to slightly opalescent, colourless to pale brown, sterile solution for intravenous (IV) infusion. Abevmy is not formulated for intravitreal use (see Section 4.4 Special Warnings and Precautions for Use, Severe eye infections following compounding for unapproved intravitreal use).
4 Clinical Particulars
4.1 Therapeutic Indications
Metastatic colorectal cancer. Abevmy (bevacizumab) in combination with fluoropyrimidine-based chemotherapy is indicated for the treatment of patients with metastatic colorectal cancer.
Locally recurrent or metastatic breast cancer. Abevmy (bevacizumab) in combination with paclitaxel is indicated for the first-line treatment of metastatic breast cancer in patients in whom an anthracycline-based therapy is contraindicated (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Advanced, metastatic or recurrent non-squamous non-small cell lung cancer (NSCLC). Abevmy (bevacizumab), in combination with carboplatin and paclitaxel, is indicated for first-line treatment of patients with unresectable advanced, metastatic or recurrent, non-squamous, non-small cell lung cancer.
Advanced and/or metastatic renal cell cancer. Abevmy (bevacizumab) in combination with interferon alfa-2a is indicated for treatment of patients with advanced and/or metastatic renal cell cancer.
Grade IV glioma. Abevmy (bevacizumab) as a single agent, is indicated for the treatment of patients with Grade IV glioma after relapse or disease progression after standard therapy, including chemotherapy.
Epithelial ovarian, fallopian tube or primary peritoneal cancer. Abevmy (bevacizumab) in combination with carboplatin and paclitaxel, is indicated for first-line treatment of patients with advanced (FIGO stages IIIB, IIIC and IV) epithelial ovarian, fallopian tube, or primary peritoneal cancer.
Recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer. Abevmy (bevacizumab) in combination with carboplatin and paclitaxel or in combination with carboplatin and gemcitabine, is indicated for the treatment of patients with first recurrence of platinum-sensitive, epithelial ovarian, fallopian tube, or primary peritoneal cancer who have not received prior bevacizumab or other VEGF-targeted angiogenesis inhibitors.
Abevmy (bevacizumab) in combination with paclitaxel, topotecan or pegylated liposomal doxorubicin is indicated for the treatment of patients with recurrent, platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer who have received no more than two prior chemotherapy regimens and have not received any prior anti-angiogenic therapy including bevacizumab.
Cervical cancer. Abevmy (bevacizumab) in combination with paclitaxel and cisplatin is indicated for the treatment of persistent, recurrent or metastatic carcinoma of the cervix. Abevmy (bevacizumab) in combination with paclitaxel and topotecan is an acceptable alternative where cisplatin is not tolerated or not indicated.
4.2 Dose and Method of Administration
In order to improve traceability of biological medicinal products, the trade name and the batch number of the administered product should be clearly recorded in the patient dispensing record.
Abevmy should be administered under the supervision of a physician experienced in the use of antineoplastic medicinal products.
Recommended dose. Metastatic colorectal cancer. The recommended dose of Abevmy, administered as an IV infusion, is as follows:
First-line treatment. 5 mg/kg of body weight given once every 2 weeks or 7.5 mg/kg of body weight given once every 3 weeks.
Second-line treatment. 10 mg/kg of body weight given every 2 weeks or 15 mg/kg of body weight given once every 3 weeks.
It is recommended that Abevmy treatment be continued until progression of the underlying disease.
Locally recurrent or metastatic breast cancer. The recommended dose of Abevmy is 10 mg/kg of body weight given once every 2 weeks or 15 mg/kg of body weight given once every 3 weeks as an IV infusion.
It is recommended that Abevmy treatment be continued until progression of the underlying disease.
Advanced, metastatic or recurrent non-squamous non-small cell lung cancer. The recommended dose of Abevmy in combination with carboplatin and paclitaxel is 15 mg/kg of body weight given once every 3 weeks as an IV infusion.
Abevmy is administered in addition to carboplatin and paclitaxel for up to 6 cycles of treatment followed by Abevmy as a single agent until disease progression.
Advanced and/or metastatic renal cell cancer. The recommended dose of Abevmy is 10 mg/kg given once every 2 weeks as an IV infusion. It is recommended that Abevmy treatment be continued until progression of the underlying disease.
Abevmy should be given in combination with IFN alfa-2a (Roferon-A). The recommended IFN alfa-2a dose is 9 MIU three times a week, however, if 9 MIU is not tolerated, the dosage may be reduced to 6 MIU and further to 3 MIU three times a week (see Section 5.1 Pharmacodynamic Properties, Clinical trials). Please also refer to the Roferon-A Product Information.
Grade IV glioma. The recommended dose of Abevmy is 10 mg/kg of body weight given once every 2 weeks or 15 mg/kg of body weight given once every 3 weeks as an IV infusion.
It is recommended that Abevmy treatment be continued until progression of the underlying disease.
Epithelial ovarian, fallopian tube or primary peritoneal cancer. The recommended dose of Abevmy administered as an IV infusion is as follows:
First line treatment. 15 mg/kg of body weight given once every 3 weeks in combination with carboplatin and paclitaxel for up to 6 cycles of treatment, followed by continued use of Abevmy as single agent.
It is recommended that Abevmy treatment be continued for a total of 15 months therapy or until disease progression, whichever occurs earlier.
Treatment of recurrent disease. Platinum sensitive: 15 mg/kg of body weight given once every 3 weeks in combination with carboplatin and paclitaxel for 6 cycles (up to 8 cycles) followed by continued use of Abevmy as a single agent until disease progression.
Alternatively, 15 mg/kg of body weight given once every 3 weeks in combination with carboplatin and gemcitabine for 6 cycles (up to 10 cycles), followed by continued use of Abevmy as single agent until disease progression.
Platinum resistant: 10 mg/kg body weight given once every 2 weeks when administered in combination with one of the following agents - paclitaxel or topotecan (given weekly) or pegylated liposomal doxorubicin. Alternatively, 15 mg/kg every 3 weeks when administered in combination with topotecan given on days 1-5, every 3 weeks. (See Section 5.1 Pharmacodynamic Properties, Clinical trials, Study MO22224 (AURELIA) for descriptions of the chemotherapy regimens.)
It is recommended that treatment be continued until disease progression.
Cervical cancer. Abevmy is administered in combination with paclitaxel and cisplatin or, if cisplatin is not tolerated or not indicated, paclitaxel and topotecan (see Section 5.1 Pharmacodynamic Properties, Clinical trials, Study GOG-0240 for further details on the chemotherapy regimens).
The recommended dose of Abevmy is 15 mg/kg of body weight given once every 3 weeks as an IV infusion.
It is recommended that Abevmy treatment be continued until progression of the underlying disease.
Dose reduction. Dose reduction of Abevmy for adverse reactions is not recommended. If indicated, Abevmy should either be discontinued or temporarily suspended (see Section 4.4 Special Warnings and Precautions for Use).
Special dosage instructions. Children and adolescents. The safety and efficacy of Abevmy in patients under the age of 18 years have not been established.
Elderly. No dose adjustment is required in the elderly.
Renal impairment. The safety and efficacy of Abevmy have not been studied in patients with renal impairment.
Hepatic impairment. The safety and efficacy of Abevmy have not been studied in patients with hepatic impairment.
Preparing the infusion. Abevmy should be prepared by a healthcare professional using aseptic technique. Withdraw the necessary amount of Abevmy and dilute to the required administration volume with 0.9% sodium chloride solution. The concentration of the final Abevmy solution should be kept within the range of 1.4-16.5 mg/mL.
No incompatibilities between Abevmy and polyvinyl chloride (PVC) or polyolefin (PO) bags have been observed.
Abevmy infusions should not be administered or mixed with dextrose or glucose solutions.
Method of administration. The initial Abevmy dose should be delivered over 90 minutes as an IV infusion. If the first infusion is well tolerated, the second infusion may be administered over 60 minutes. If the 60-minute infusion is well tolerated, all subsequent infusions may be administered over 30 minutes.
Do not administer as an intravenous push or bolus.
Abevmy is not formulated for intravitreal use (see Section 4.4 Special Warnings and Precautions for Use, Severe eye infections following compounding for unapproved intravitreal use).
4.3 Contraindications
Abevmy is contraindicated in patients with: known hypersensitivity to any components of the product; Chinese hamster ovary cell products or other recombinant human or humanised antibodies.
4.4 Special Warnings and Precautions for Use
Gastrointestinal perforations and fistulae. Patients may be at increased risk for the development of gastrointestinal (GI) perforation and gallbladder perforation when treated with bevacizumab. Bevacizumab should be permanently discontinued in patients who develop GI perforation. Patients treated with bevacizumab for persistent, recurrent, or metastatic cervical cancer may be at increased risk of fistulae between the vagina and any part of the GI tract (GI-vaginal fistulae).
Bevacizumab has been associated with serious cases of GI perforation. GI perforations have been reported in clinical trials with an incidence of < 1% in patients with metastatic breast cancer or NSCLC, up to 2% in patients with metastatic colorectal cancer or ovarian cancer (first-line treatment), and up to 2.7% in patients with metastatic colorectal cancer (including GI fistula and abscess). Cases of GI perforations have also been observed in patients with relapsed glioblastoma. Fatal outcome was reported in approximately a third of serious cases of GI perforations, which represents between 0.2%-1% of all bevacizumab-treated patients.
Patients treated for recurrent platinum-resistant ovarian cancer should not have a history or symptoms of bowel obstruction, abdominal fistulae or clinical or radiological evidence of recto-sigmoid involvement. Patient eligibility in the pivotal study MO22224 was also limited to those with two or fewer prior chemotherapy regimens.
From a clinical trial in patients with persistent, recurrent, or metastatic cervical cancer (Study GOG-0240), GI perforations (all Grades) were reported in 3.2% of patients, all of whom had a history of prior pelvic radiation. The incidence of GI-vaginal fistulae was 8.3% in bevacizumab-treated patients and 0.9% in control patients, all of whom had a history of prior pelvic radiation. Patients who develop GI-vaginal fistulae may also have bowel obstructions and require surgical intervention as well as diverting ostomies.
In other bevacizumab clinical trials, GI fistulae (all Grades) have been reported with an incidence of up to 2% in patients with metastatic colorectal cancer and ovarian cancer but were also reported less commonly in patients with other types of cancer. The occurrence of those events varied in type and severity, ranging from free air seen on the plain abdominal X-ray, which resolved without treatment, to intestinal perforation with abdominal abscess and fatal outcome. In some cases, underlying intra-abdominal inflammation was present, either from gastric ulcer disease, tumour necrosis, diverticulitis or chemotherapy-associated colitis. A causal association of intra-abdominal inflammatory process and GI perforation to bevacizumab has not been established.
Hypertension. An increased incidence of hypertension was observed in patients treated with bevacizumab. Clinical safety data suggest that the incidence of hypertension is likely to be dose-dependent. Pre-existing hypertension should be adequately controlled before starting bevacizumab treatment. There is no information on the effect of bevacizumab in patients with uncontrolled hypertension at the time of initiating bevacizumab therapy. Monitoring of blood pressure is recommended during bevacizumab therapy.
In most cases hypertension was controlled adequately using standard anti-hypertensive treatment appropriate for the individual situation of the affected patient. Bevacizumab should be permanently discontinued if medically significant hypertension cannot be adequately controlled with antihypertensive therapy, or if, the patient develops hypertensive crisis or hypertensive encephalopathy (see Section 4.8 Adverse Effects (Undesirable Effects), Post-marketing experience).
An increased incidence of hypertension (all grades) of up to 42.1% has been observed in patients treated with bevacizumab compared with up to 14% in the comparator arm. In clinical trials across all indications the overall incidence of Grade 3-4 hypertension in patients receiving bevacizumab ranged from 0.4% to 17.9%. Grade 4 hypertension (hypertensive crisis) occurred in up to 1.0% of patients treated with bevacizumab compared to up to 0.2% patients treated with the same chemotherapy alone.
Hypertension was generally treated with oral anti-hypertensives such as angiotensin-converting enzyme inhibitors, diuretics and calcium-channel blockers. It rarely resulted in discontinuation of bevacizumab treatment or hospitalisation. The use of diuretics to manage hypertension is not advised in patients who receive a cisplatin-based chemotherapy regimen.
Very rare cases of hypertensive encephalopathy have been reported, some of which were fatal (see Section 4.8 Adverse Effects (Undesirable Effects), Post-marketing experience). The risk of bevacizumab-associated hypertension did not correlate with the patients' baseline characteristics, underlying disease or concomitant therapy.
Wound healing. Bevacizumab may adversely affect the wound healing process; bevacizumab therapy should not be initiated for at least 28 days following major surgery or until the surgical wound is fully healed. In patients who experience wound healing complications during bevacizumab therapy, bevacizumab should be withheld until the wound is fully healed. Bevacizumab therapy should be withheld for elective surgery.
Across metastatic colorectal cancer clinical trials there was no increased risk of post-operative bleeding or wound healing complications observed in patients who underwent major surgery between 28-60 days prior to starting bevacizumab therapy. An increased incidence of post-operative bleeding or wound healing complications occurring within 60 days of major surgery was observed if the patient was being treated with bevacizumab at the time of surgery. The incidence varied between 10% (4/40) and 20% (3/15).
Serious wound healing complications, including anastomotic complications, have been reported, some of which had a fatal outcome.
In locally recurrent and metastatic breast cancer, National Cancer Institute-Common Toxicity Criteria (NCI-CTC) Grade 3-5 wound healing complications were observed in up to 1.1% of patients receiving bevacizumab compared with up to 0.9% of patients in the control arms.
In Study AVF3708g, patients with relapsed GBM, the incidence of post-operative wound healing complications (craniotomy site wound dehiscence and cerebrospinal fluid leak) was 3.6% in patients treated with single-agent bevacizumab and 1.3% in patients treated with bevacizumab and irinotecan.
Necrotising fasciitis including fatal cases, has rarely been reported in patients treated with bevacizumab; usually secondary to wound healing complications, gastrointestinal perforation or fistula formation. Bevacizumab therapy should be discontinued in patients who develop necrotising fasciitis, and appropriate treatment should be promptly initiated (see Section 4.8 Adverse Effects (Undesirable Effects), Post-marketing experience).
Thromboembolism. Arterial thromboembolic events. An increased incidence of arterial thromboembolic events has been observed in patients treated with bevacizumab across indications including cerebrovascular accidents, myocardial infarction, transient ischaemic attacks, and other arterial thromboembolic events.
In clinical trials, the overall incidence ranged up to 5.9% in the bevacizumab-containing arms compared with up to 1.7% in the chemotherapy control arms. Fatal outcome was reported in 0.8% of patients receiving bevacizumab in combination with chemotherapy compared to 0.5% of patients receiving chemotherapy alone. Cerebrovascular accidents (including transient ischaemic attacks) were reported in up to 2.3% of bevacizumab-treated patients versus 0.5% of patients in the control group. Myocardial infarction was reported in 1.4% of bevacizumab treated versus 0.7% of patients in the observed control group. In the uncontrolled study AVF3708g, in patients with relapsed GBM, arterial thromboembolic events were observed in 6.3% (5/79) of patients who received bevacizumab in combination with irinotecan compared to 4.8% (4/84) of patients who received bevacizumab alone. Bevacizumab is approved for the treatment of relapsed GBM as a single agent. Bevacizumab, in combination with fluoropyrimidine-based chemotherapy (5-fluorouracil ± irinotecan), is indicated only for the treatment of patients with metastatic colorectal cancer.
Bevacizumab should be permanently discontinued in patients who develop arterial thromboembolic events.
Patients receiving bevacizumab plus chemotherapy with a history of arterial thromboembolism, diabetes or age greater than 65 years have an increased risk of developing arterial thromboembolic events during bevacizumab therapy. Caution should be taken when treating such patients with bevacizumab.
Venous thromboembolic events. In clinical trials across indications, the overall incidence of venous thromboembolic events ranged from 2.8% to 17.3% in the bevacizumab containing arms compared to 3.2% to 15.6% in the chemotherapy control arms. Venous thromboembolic events include deep venous thrombosis and pulmonary embolism.
Patients may be at risk of developing venous thromboembolic events, including pulmonary embolism under bevacizumab treatment. Patients treated with bevacizumab for persistent, recurrent, or metastatic cervical cancer may be at increased risk of venous thromboembolic events, compared to patients receiving chemotherapy alone.
Bevacizumab should be discontinued in patients with life-threatening (Grade 4) venous thromboembolic events, including pulmonary embolism. Patients with thromboembolic events ≤ Grade 3 need to be closely monitored.
Grade 3-5 venous thromboembolic events have been reported in up to 10.6% of patients treated with chemotherapy plus bevacizumab compared with up to 5.4% in patients with chemotherapy alone. Patients who have experienced a venous thromboembolic event may be at higher risk for a recurrence if they receive bevacizumab in combination with chemotherapy versus chemotherapy alone.
Haemorrhage. Patients treated with bevacizumab have an increased risk of haemorrhage, especially tumour-associated haemorrhage. Bevacizumab should be permanently discontinued in patients who experience Grade 3 or 4 bleeding during bevacizumab therapy.
In clinical trials across all indications the overall incidence of NCI-CTC Grade 3-5 bleeding events ranged from 0.4% to 6.9% in bevacizumab-treated patients, compared to 0 to 4.5% of patients in the chemotherapy control group. Haemorrhagic events observed in bevacizumab clinical trials were predominantly tumour-associated haemorrhage and minor mucocutaneous haemorrhage (e.g. epistaxis).
Patients with untreated central nervous system (CNS) metastases have been routinely excluded from clinical studies with bevacizumab, based on imaging procedures or signs and symptoms. However, 2 studies of bevacizumab in ovarian cancer provide a comparison with standard carboplatin/paclitaxel therapy of the incidence of CNS and non-CNS haemorrhage in patients without cerebral metastases. In Study GOG-0218, three patients who received extended treatment with bevacizumab developed CNS haemorrhage, with 1 death, and the same number in the bevacizumab arm of Study BO17707, also with 1 death. No CNS haemorrhage occurred in the control arms. Non-CNS haemorrhages were observed in Study GOG-0218 in 15.9% of control patients vs. 35.7% and 37.0% in the short and extended duration bevacizumab arms; in B017707 they were observed in 11% of control patients and 39.4% of the bevacizumab-treated patients. Most of the non-CNS haemorrhages were Grade 3 or less (GOG-0218: three events in the bevacizumab arm were Grade 4; B017707: one patient in the bevacizumab arm had a Grade 4 event and 2 patients in the control arm had a Grade 4 or higher event, one Grade 4 event and one Grade 5 event). In a third study of ovarian cancer (MO22224) one patient in the CT + bevacizumab arm experienced a Grade 4 GI haemorrhage which was ongoing at the time of death, and one patient who had crossed over to bevacizumab monotherapy died from a Grade 5 GI haemorrhage.
Patients should be monitored for signs and symptoms of CNS bleeding, and bevacizumab treatment discontinued in case of intracranial bleeding.
There is no information on the safety profile of bevacizumab in patients with congenital bleeding diathesis, acquired coagulopathy or in patients receiving full dose of anticoagulants for the treatment of thromboembolism prior to starting bevacizumab therapy, as such patients were excluded from clinical trials. Therefore, caution should be exercised before initiating bevacizumab therapy in these patients. However, patients who developed venous thrombosis while receiving bevacizumab therapy did not appear to have an increased rate of Grade 3 or above bleeding when treated with full dose of warfarin and bevacizumab concomitantly.
Tumour-associated haemorrhage. Major or massive pulmonary haemorrhage/haemoptysis has been observed primarily in studies in patients with NSCLC. Possible risk factors include squamous cell histology, treatment with antirheumatic/anti-inflammatory drugs, treatment with anticoagulants, prior radiotherapy, bevacizumab therapy, previous medical history of atherosclerosis, central tumour location and cavitation of tumours prior to or during therapy. The only variables that showed statistically significant correlations with bleeding were bevacizumab therapy and squamous cell histology. Patients with NSCLC of known squamous cell histology or mixed cell type with predominant squamous cell histology were excluded from subsequent studies, while patients with unknown tumour histology were included.
In patients with NSCLC excluding predominant squamous histology, all grade events were seen with a frequency of up to 9% when treated with bevacizumab plus chemotherapy compared with 5% in the patients treated with chemotherapy alone. Grade 3-5 events have been observed in up to 2.3% of patients treated with bevacizumab plus chemotherapy as compared with < 1% with chemotherapy alone. Major or massive pulmonary haemorrhage/haemoptysis can occur suddenly and up to two thirds of the serious pulmonary haemorrhages resulted in a fatal outcome.
GI haemorrhages, including rectal bleeding and melaena have been reported in colorectal patients, and have been assessed as tumour-associated haemorrhages.
Tumour-associated haemorrhages have also been seen rarely in other tumour types and locations and include cases of CNS bleeding in patients with CNS metastases and glioblastoma (GBM). In an exploratory retrospective analysis of data from 13 completed randomised trials in patients with various tumour types, 3 patients out of 91 (3.3%) with brain metastases experienced CNS bleeding (all Grade 4) when treated with bevacizumab, compared to 1 case (Grade 5) out of 96 patients (1%) that were not exposed to bevacizumab. In 2 subsequent studies in patients with treated brain metastases (approx. 800 patients treated with bevacizumab), 1 case of Grade 2 CNS haemorrhage was reported. One patient in the CT + bevacizumab arm of the recurrent platinum resistant ovarian cancer study MO22224 experienced a Grade 3 haemorrhagic ascites which subsequently resolved.
Intracranial haemorrhage can occur in patients with relapsed GBM. In study AVF3708g, CNS haemorrhage was reported in 2.4% (2/84) of patients in the single-agent bevacizumab arm (Grade 1) and in 3.8% (3/79) of patients treated with bevacizumab and irinotecan (Grades 1, 2 and 4).
Mucocutaneous haemorrhage. Mucocutaneous haemorrhages were seen in up to 50% of patients treated with bevacizumab, across all bevacizumab clinical trials. These were most commonly NCI-CTC Grade 1 epistaxis that lasted < 5 minutes, resolved without medical intervention and did not require any changes in bevacizumab treatment regimen. Clinical safety data suggest that the incidence of minor mucocutaneous haemorrhage (e.g. epistaxis) may be dose-dependent. There have been less common events of minor mucocutaneous haemorrhage in other locations such as gingival bleeding or vaginal bleeding.
Pulmonary haemorrhage/haemoptysis. Patients with NSCLC treated with bevacizumab may be at risk for serious, and in some cases fatal, pulmonary haemorrhage/haemoptysis. Patients with recent pulmonary haemorrhage/haemoptysis (> ½ teaspoon red blood) should not be treated with bevacizumab.
Aneurysms and artery dissections. The use of VEGF pathway inhibitors in patients with or without hypertension may promote the formation of aneurysms and/or artery dissections. Before initiating bevacizumab, this risk should be carefully considered in patients with risk factors such as hypertension or history of aneurysm.
Posterior reversible encephalopathy syndrome (PRES). There have been rare reports of bevacizumab-treated patients developing signs and symptoms that are consistent with posterior reversible encephalopathy syndrome (PRES) (see Section 4.8 Adverse Effects (Undesirable Effects), Post-marketing experience), a rare neurological disorder, which can present with the following signs and symptoms among others: seizures, headache, altered mental status, visual disturbance, or cortical blindness, with or without associated hypertension.
Two confirmed cases (0.8%) of PRES were reported in Study AVF4095g (OCEANS). Symptoms usually resolved or improved within days, although some patients experienced neurologic sequelae.
Two confirmed cases of PRES were reported in Study MO22224 (AURELIA). One case occurred during concurrent administration of CT + bevacizumab and the other after cross-over from CT to bevacizumab monotherapy.
A diagnosis of PRES requires confirmation by brain imaging, preferably magnetic resonance imaging (MRI). In patients developing PRES, treatment of specific symptoms including control of hypertension is recommended along with discontinuation of bevacizumab. The safety of reinitiating bevacizumab therapy in patients previously experiencing PRES is not known.
Proteinuria. Patients with a history of hypertension may be at increased risk for the development of proteinuria when treated with bevacizumab. There is evidence suggesting that all Grade proteinuria may be dose-dependent. Testing for proteinuria is recommended prior to the start of bevacizumab therapy. In most clinical studies urine protein levels of ≥ 2 g/24 h led to the holding of bevacizumab until recovery to < 2 g/24 h.
In clinical trials, the incidence of proteinuria was higher in patients receiving bevacizumab in combination with chemotherapy compared to those who received chemotherapy alone. Grade 4 proteinuria was common in patients treated with bevacizumab.
Proteinuria has been reported within the range of 0.7% to 38% of patients receiving bevacizumab. Proteinuria ranged in severity from clinically asymptomatic, transient, trace proteinuria to nephrotic syndrome. Grade 3 proteinuria was reported in up to 8.1% of treated patients. Grade 4 proteinuria (nephrotic syndrome) was seen in up to 1.4% of treated patients. In the event of nephrotic syndrome or isolated Grade 4 proteinuria, bevacizumab should be permanently discontinued.
Congestive heart failure. Caution should be exercised when treating patients with clinically significant cardiovascular disease or pre-existing congestive heart failure (CHF).
Prior anthracyclines exposure and/or prior radiation to the chest wall may be possible risk factors for the development of CHF.
Events consistent with CHF were reported in clinical trials in all cancer indications studied to date. The findings ranged from asymptomatic declines in left ventricular ejection fraction to symptomatic CHF, requiring treatment or hospitalisation. Most of the patients who experienced CHF had metastatic breast cancer and had received previous treatment with anthracyclines, prior radiotherapy to the left chest wall or other risk factors for CHF were present.
In phase III studies in patients with metastatic breast cancer, CHF Grade 3 or higher was reported in up to 3.5% of patients treated with bevacizumab in combination with chemotherapy compared with up to 0.9% in the control arms. Most patients who developed CHF during mBC trials showed improved symptoms and/or left ventricular function following appropriate medical therapy.
In most clinical trials of bevacizumab, patients with pre-existing CHF of NYHA II-IV were excluded, therefore, no information is available on the risk of CHF in this population.
An increased incidence of CHF has been observed in a phase III clinical trial of patients with diffuse large B-cell lymphoma when receiving bevacizumab with a cumulative doxorubicin dose greater than 300 mg/m2. This clinical trial compared rituximab / cyclophosphamide / doxorubicin / vincristine / prednisone (R-CHOP) plus bevacizumab to R-CHOP without bevacizumab. While the incidence of CHF was, in both arms, above that previously observed for doxorubicin therapy, the rate was higher in the R-CHOP plus bevacizumab arm.
Neutropenia. Increased rates of severe neutropenia, febrile neutropenia, or infection with severe neutropenia (including some fatalities) have been observed in patients treated with some myelotoxic chemotherapy regimens plus bevacizumab in comparison to chemotherapy alone (e.g. bevacizumab with platinum or taxane based chemotherapies); (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Non-GI fistulae. Patients may be at increased risk for the development of fistulae when treated with bevacizumab. Bevacizumab use has been associated with serious cases of fistulae including events resulting in death. From a clinical trial in patients with persistent, recurrent or metastatic cervical cancer (GOG-0240), 1.8% of bevacizumab-treated patients and 1.4% of control patients were reported to have had non-gastrointestinal vaginal, vesical, or female genital tract fistulae.
Uncommon (≥ 0.1% to < 1%) reports of other types of fistulae that involve areas of the body other than the GI tract (e.g. bronchopleural, biliary fistulae) were observed across various indications. Fistulae have also been reported in post-marketing experience.
Events were reported at various time points during treatment ranging from one week to greater than 1 year from initiation of bevacizumab, with most events occurring within the first 6 months of therapy. Permanently discontinue bevacizumab in patients with tracheo-oesophageal fistula or any Grade 4 fistula. Limited information is available on the continued use of bevacizumab in patients with other fistulae. In cases of internal fistula not arising in the GI tract, discontinuation of bevacizumab should be considered.
Hypersensitivity reactions (including anaphylactic shock)/ infusion reactions. In some clinical trials anaphylactic and anaphylactoid-type reactions were reported more frequently in patients receiving bevacizumab in combination with chemotherapies than with chemotherapy alone. The incidence of these reactions in some clinical trials of bevacizumab is common (up to 5% in bevacizumab-treated patients).
Patients may be at risk of developing infusion/hypersensitivity reactions (including anaphylactic shock, see Section 4.8 Adverse Effects (Undesirable Effects)). Close observation of the patient during and following the administration of bevacizumab is recommended as expected for any infusion of a therapeutic humanised monoclonal antibody. If a reaction occurs, the infusion should be discontinued, and appropriate medical therapies should be administered. A systematic premedication is not warranted.
Severe eye infections following compounding for unapproved intravitreal use. Individual cases and clusters of serious ocular adverse events have been reported (including infectious endophthalmitis and other ocular inflammatory conditions) following unapproved intravitreal use of bevacizumab compounded from vials approved for intravenous administration in cancer patients. Some of these events have resulted in various degrees of visual loss, including permanent blindness (see Section 4.8 Adverse Effects (Undesirable Effects), Post-marketing experience).
Systemic effects following intravitreal use. A reduction of circulating VEGF concentration has been demonstrated following intravitreal anti-VEGF therapy. Systemic adverse reactions including non-ocular haemorrhages and arterial thromboembolic reactions have been reported following intravitreal injection of VEGF inhibitors.
Osteonecrosis of the jaw (ONJ). Cases of ONJ have been reported in cancer patients treated with bevacizumab (see Section 4.8 Adverse Effects (Undesirable Effects), Post-marketing experience). Most had received prior or concomitant intravenous bisphosphonates, for which ONJ is an identified risk. Caution should be exercised when Abevmy and intravenous bisphosphonates are administered simultaneously or sequentially. Invasive dental procedures are also an identified risk factor. A dental examination and appropriate preventive dentistry should be considered prior to starting bevacizumab. In patients who have previously received or are receiving intravenous bisphosphonates invasive dental procedures should be avoided, if possible.
Use in the elderly. In randomised clinical trials, age > 65 years was associated with an increased risk of developing arterial thromboembolic events including cerebrovascular accidents, transient ischaemic attacks and myocardial infarction, as compared to those aged ≤ 65 years when treated with bevacizumab. Other reactions with a higher frequency seen in patients over 65 were Grade 3-4 leucopenia and thrombocytopenia; and all grade neutropenia, diarrhoea, nausea, headache and fatigue.
In study MO22224 alopecia, mucosal inflammation, peripheral sensory neuropathy, proteinuria and hypertension were observed at a higher incidence in elderly patients (≥ 65 years) receiving CT + bevacizumab compared to those aged < 65 years treated with CT + bevacizumab. Serious adverse events also occurred at a higher incidence in patients ≥ 65 years treated with CT + bevacizumab (38.6%) compared to patients < 65 years (26.6%).
From a clinical trial in patients with metastatic colorectal cancer (study AVF2107) no increase in the incidence of other reactions including GI perforation, wound healing complications, congestive heart failure and haemorrhage, was observed in elderly patients (> 65 years) receiving bevacizumab as compared to those aged ≤ 65 years treated with bevacizumab.
Paediatric use. Bevacizumab is not approved for use in patients under the age of 18 years. The safety and effectiveness of bevacizumab in this population have not been established. Addition of bevacizumab to standard of care did not demonstrate clinical benefit in paediatric patients in two phase II clinical trials: one in paediatric high grade glioma and one in paediatric metastatic rhabdomyosarcoma or non-rhabdomyosarcoma soft tissue sarcoma. In published reports, cases of osteonecrosis at sites other than the jaw have been observed in patients under the age of 18 years exposed to bevacizumab.
In a 26 week pre-clinical study in cynomolgus monkeys, physeal dysplasia was observed in young animals with open growth plates, at bevacizumab average serum concentrations below the expected human therapeutic average serum concentrations.
Effects on laboratory tests. See Section 4.8 Adverse Effects (Undesirable Effects), Laboratory abnormalities.
4.5 Interactions with Other Medicines and Other Forms of Interactions
Effect of antineoplastic agents on bevacizumab pharmacokinetics. No clinically relevant pharmacokinetic interaction of co-administered chemotherapy on bevacizumab pharmacokinetics has been observed based on the results of a population pharmacokinetic analysis. There was neither statistical significance nor clinically relevant difference in clearance of bevacizumab in patients receiving bevacizumab monotherapy compared to patients receiving bevacizumab in combination with IFN alfa-2a or other chemotherapies (IFL, 5-FU/LV, carboplatin-paclitaxel, capecitabine, doxorubicin or cisplatin/gemcitabine).
Effect of bevacizumab on the pharmacokinetics of other antineoplastic agents. Results from a drug-drug interaction study, AVF3135g, demonstrated no significant effect of bevacizumab on the pharmacokinetics of irinotecan and its active metabolite SN38.
Results from study NP18587 demonstrated no significant effect of bevacizumab on the pharmacokinetic of capecitabine and its metabolites, and on the pharmacokinetics of oxaliplatin, as determined by measurement of free and total platinum.
Results from study B017705 demonstrated no significant effect of bevacizumab on the pharmacokinetics of IFN alfa-2a.
Combination of bevacizumab and sunitinib malate. In two clinical studies of metastatic renal cell carcinoma, microangiopathic haemolytic anaemia (MAHA) was reported in 7/19 patients treated with bevacizumab (10 mg/kg every two weeks) and sunitinib malate (50 mg daily) combination.
MAHA is a haemolytic disorder which can present with red cell fragmentation, anaemia, and thrombocytopenia. In addition, hypertension (including hypertensive crisis), elevated creatinine, and neurological symptoms were observed in some of these patients. All of these findings were reversible upon discontinuation of bevacizumab and sunitinib malate (see Section 4.4 Special Warnings and Precautions for Use, Hypertension, Proteinuria and Posterior reversible encephalopathy syndrome (PRES)).
Combination of bevacizumab with pegylated liposomal doxorubicin (PLD), platinum- or taxane-based chemotherapies. Increased rates of severe neutropenia, febrile neutropenia, or infection with severe neutropenia (including some fatalities) have been observed in patients treated with some myelotoxic chemotherapy regimens plus bevacizumab in comparison to chemotherapy alone (e.g. bevacizumab with platinum- or taxane-based chemotherapies). Bevacizumab may also exacerbate other adverse reactions commonly seen with chemotherapy when combined with chemotherapeutic agents (see Section 4.8 Adverse Effects (Undesirable Effects)).
Radiotherapy. The safety and efficacy of concomitant administration of radiotherapy and bevacizumab have not been established.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. Bevacizumab may impair female fertility, therefore fertility preservation strategies should be discussed with women of child-bearing potential prior to starting treatment with bevacizumab.
No specific studies in animals have been performed to evaluate the effect of bevacizumab on fertility. No adverse effect on the male reproductive organ was observed in repeat dose toxicity studies in cynomolgus monkeys, but inhibition of ovarian function was observed in females. This was characterised by decreases in ovarian and/or uterine weight and the number of corpora lutea, a reduction in endometrial proliferation and an inhibition of follicular maturation in cynomolgus monkeys treated with bevacizumab. The lowest dose tested in the 26 week study (2 mg/kg weekly which corresponds to 0.6-fold the human therapeutic dose based on AUC) caused a reduction in uterine weight, however the reduction was not statistically significant. In rabbits, administration of 50 mg/kg of bevacizumab IV for 3 or 4 doses every 4 days resulted in decreases in ovarian and/or uterine weight and number of corpora lutea. The changes in both monkeys and rabbits were reversible upon cessation of treatment.
The incidence of new cases of ovarian failure (defined as amenorrhoea lasting 3 or more months, FSH level ≥ 30 mIU/mL and a negative serum β-HCG pregnancy test) was evaluated in a sub-study of 295 premenopausal women, treated with or without bevacizumab in adjuvant colon cancer therapy. New cases of ovarian failure were reported more frequently in patients receiving bevacizumab (39% in the bevacizumab group compared to 2.6% in the control). After discontinuation of bevacizumab, ovarian function recovered in a majority of women. Long term effects of treatment with bevacizumab on fertility are unknown, however a higher rate of long-term amenorrhoea following bevacizumab therapy was observed in patients < 40 years (40% amenorrhoeic at 24 months after bevacizumab treatment compared to 6% in the control group).
Use in pregnancy. (Category D)
There are no adequate and well-controlled studies in pregnant women. IgGs are known to cross the placental barrier, and bevacizumab may inhibit angiogenesis in the foetus. Angiogenesis has been shown to be critically important to fetal development. The inhibition of angiogenesis following administration of bevacizumab could result in an adverse outcome of pregnancy. Therefore, bevacizumab should not be used during pregnancy. In the post-marketing setting, cases of fetal abnormalities in women treated with bevacizumab alone or in combination with known embryotoxic chemotherapeutics have been observed (see Section 4.8 Adverse Effects (Undesirable Effects), Post-marketing experience).
In women with childbearing potential, appropriate contraceptive measures should be used during bevacizumab therapy. Based on pharmacokinetic considerations, contraceptive measures should be used for at least 6 months following the last dose of bevacizumab.
Bevacizumab has been shown to be embryotoxic and teratogenic when administered to rabbits. Observed effects included decreases in fetal body weights, an increased number of fetal resorptions and an increased incidence of specific gross and skeletal fetal alterations. Adverse fetal outcomes were observed at all tested doses. At the lowest dose tested, maternal serum AUC values were about 0.7-fold those observed in humans at the recommended clinical dose.
Use in lactation. Immunoglobulins are excreted in milk, although there are no data specifically for bevacizumab excretion in milk. Since bevacizumab could harm infant growth and development, women should be advised to discontinue breastfeeding during bevacizumab therapy and not to breast feed for at least 6 months following the last dose of bevacizumab.
4.7 Effects on Ability to Drive and Use Machines
No studies on the effects on the ability to drive and use machines have been performed. However, there is no evidence that bevacizumab treatment results in an increase in adverse events that might lead to impairment of the ability to drive or operate machinery or impairment of mental ability.
The effects of this medicine on a person's ability to drive and use machines were not assessed as part of its registration.
4.8 Adverse Effects (Undesirable Effects)
Experience from clinical trials. Clinical trials have been conducted in approximately 5,500 patients with various malignancies treated with bevacizumab, predominantly in combination with chemotherapy. The safety profile from the clinical trial population is presented in this section.
The most serious adverse drug reactions were:
Gastrointestinal perforations (see Section 4.4 Special Warnings and Precautions for Use).
Haemorrhage including pulmonary haemorrhage/haemoptysis, which is more common in NSCLC patients (see Section 4.4 Special Warnings and Precautions for Use).
Arterial and venous thromboembolism (see Section 4.4 Special Warnings and Precautions for Use).
Analyses of the clinical safety data suggest that the occurrence of hypertension and proteinuria with bevacizumab therapy are likely to be dose-dependent (see Section 4.4 Special Warnings and Precautions for Use).
The most frequently observed adverse drug reactions across clinical trials in patients receiving bevacizumab were hypertension, fatigue or asthenia, diarrhoea and abdominal pain.
Table 1 lists adverse drug reactions associated with the use of bevacizumab in combination with different chemotherapy regimens in multiple indications. These reactions had occurred either with at least a 2% difference compared to the control arm (NCI-CTC Grade 3-5 reactions) or with at least a 10% difference compared to the control arm (NCI-CTC Grade 1-5 reactions), in at least one of the major clinical trials. The adverse drug reactions listed in Table 1 fall into the following categories: very common (≥ 10%) and common (≥ 1% - < 10%). Adverse drug reactions have been included in the appropriate category according to the highest incidence seen in any of the major clinical trials. Within each frequency grouping adverse drug reactions are presented in order of decreasing seriousness. Some of the adverse reactions are reactions commonly seen with chemotherapy however bevacizumab may exacerbate these reactions when combined with chemotherapeutic agents. Examples include palmar-plantar erythrodysaesthesia syndrome with pegylated liposomal doxorubicin or capecitabine, peripheral sensory neuropathy with paclitaxel or oxaliplatin, and nail disorders or alopecia with paclitaxel.
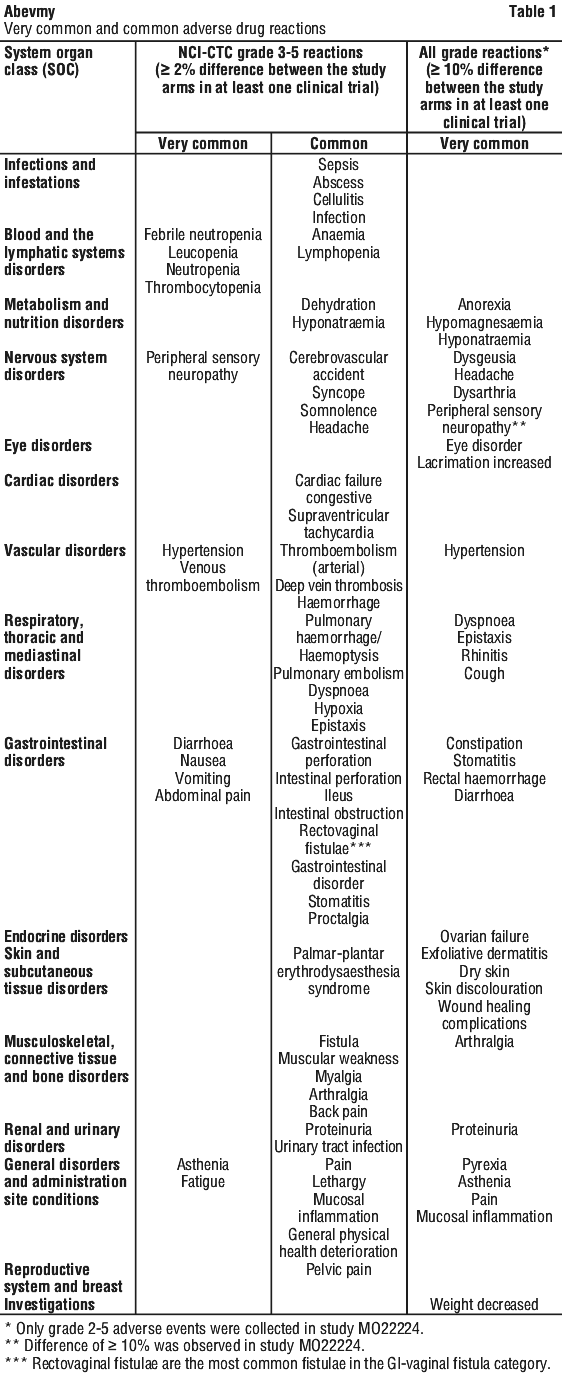
Across clinical trials, the following Grade 3 and 4 laboratory abnormalities were seen with an increased (≥ 2%) incidence in patients treated with bevacizumab compared to those in the control groups: hyperglycaemia, decreased haemoglobin, hypokalaemia, hyponatraemia, decreased white blood cell count, increased prothrombin time and normalised ratio.
Clinical trials have shown that transient increases in serum creatinine (ranging between 1.5-1.9 times baseline level), both with and without proteinuria, are associated with the use of bevacizumab. The observed increase in serum creatinine was not associated with a higher incidence of clinical manifestations of renal impairment in patients treated with bevacizumab.
Post-marketing experience. The following adverse drug reactions have been identified from post-marketing experience with bevacizumab (Table 2) based on spontaneous case reports and literature cases. Adverse drug reactions are listed according to system organ classes in MedDRA and the corresponding frequency category estimation for each adverse drug reaction is based on the following convention: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000).
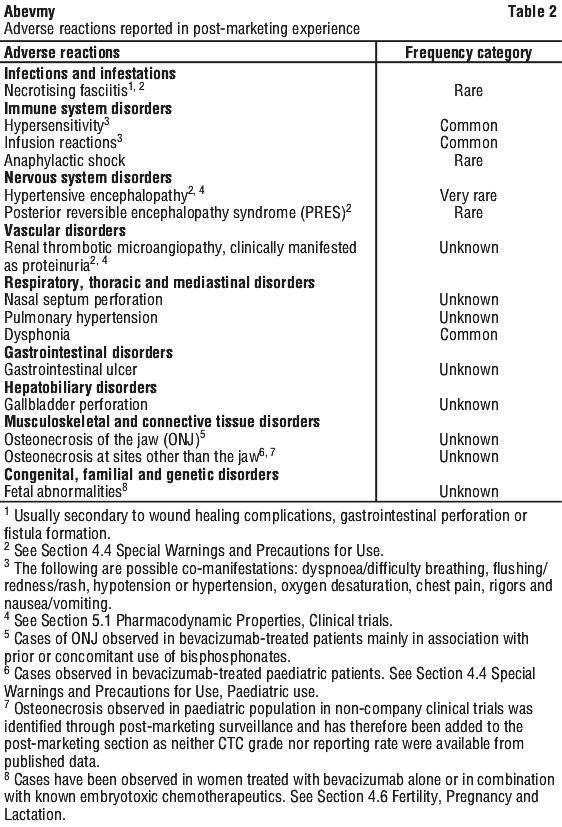
Systemic events (reported from unapproved intravitreal use). Increased risk for haemorrhagic stroke (see Section 4.4 Special Warnings and Precautions for Use); increased risk for overall mortality; increased risk for serious systemic adverse events, most of which resulted in hospitalisation (adjusted risk ratio 1.29; 95% CI: 1.01, 1.66) (Incidence 24.1%; comparator 19.0%).
Vascular disorders. Cases of aneurysms and artery dissections, sometimes fatal, have been reported with VEGFR pathway inhibitors.
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
4.9 Overdose
The highest dose tested in humans (20 mg/kg body weight, IV) was associated with severe migraine in several patients.
Treatment of overdose should consist of general supportive measures.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action. Abevmy is an antineoplastic agent containing the active ingredient, bevacizumab. Bevacizumab is a recombinant humanised monoclonal antibody that selectively binds to and neutralises the biologic activity of human vascular endothelial growth factor (VEGF). Bevacizumab contains human framework regions with antigen binding regions of a humanised murine antibody that binds to VEGF. Bevacizumab is produced by recombinant DNA technology in a Chinese hamster ovary mammalian cell expression system in a nutrient medium containing the antibiotic gentamicin and is purified by a process that includes specific viral inactivation and removal steps. Gentamicin is detectable in the final product at ≤ 0.35 ppm.
Bevacizumab inhibits the binding of VEGF to its receptors, Flt-1 and KDR, on the surface of endothelial cells. Neutralising the biologic activity of VEGF reduces the vascularisation of tumours, thereby inhibiting tumour growth. Administration of bevacizumab or its parental murine antibody to xenotransplant models of cancer in nude mice resulted in extensive anti-tumour activity in human cancers, including colon, breast, pancreas and prostate. Metastatic disease progression was inhibited, and microvascular permeability was reduced.
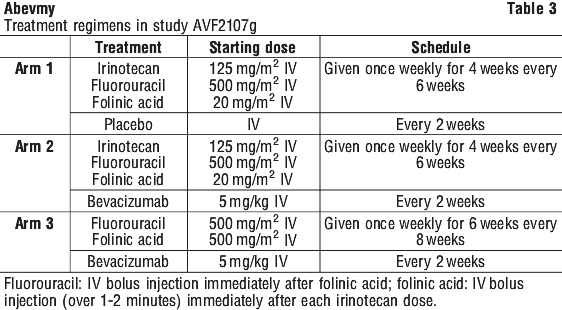
Clinical trials. Metastatic colorectal cancer. The safety and efficacy of bevacizumab in metastatic colorectal cancer were studied in two randomised, active-controlled clinical trials. Bevacizumab was combined with two chemotherapy regimens:
AVF2107g: A weekly schedule of irinotecan/bolus fluorouracil/leucovorin* (IFL) for a total of 4 weeks of each 6 week cycle.
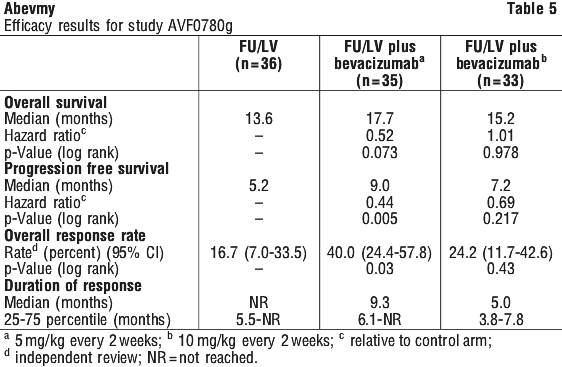
AVF0780g: In combination with bolus fluorouracil/leucovorin* (FU/LV) for a total of 6 weeks of each 8 week cycle (Roswell Park regimen).
Two additional studies were conducted in first (NO16966) and second line (E3200) treatment of metastatic carcinoma of the colon or rectum, with bevacizumab administered in the following dosing regimens, in combination with FOLFOX-4 (FU/LV/oxaliplatin) and XELOX (capecitabine/oxaliplatin):
NO16966: Bevacizumab 7.5 mg/kg of body weight every 3 weeks in combination with oral capecitabine and IV oxaliplatin (XELOX) or bevacizumab 5 mg/kg every 2 weeks in combination with leucovorin* plus fluorouracil bolus, followed by fluorouracil infusion, with IV oxaliplatin (FOLFOX-4).
E3200: Bevacizumab 10 mg/kg of body weight every 2 weeks in combination with leucovorin* and fluorouracil bolus, followed by fluorouracil infusion, with IV oxaliplatin (FOLFOX-4).
* The Australian Approved Name for leucovorin is folinic acid.
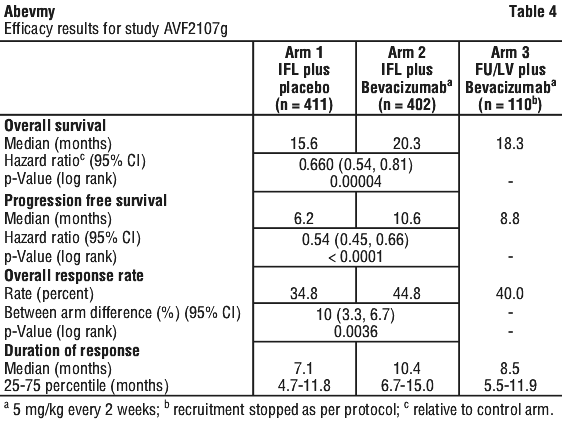
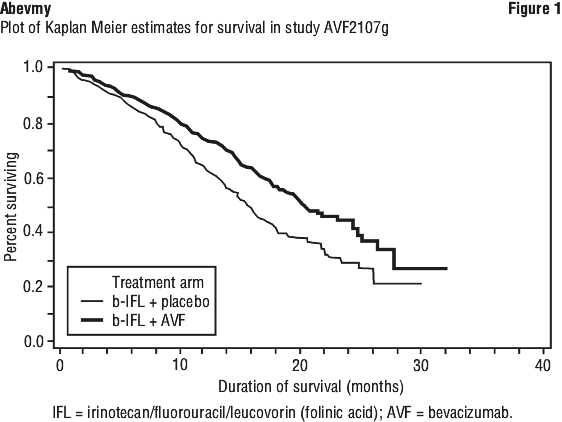
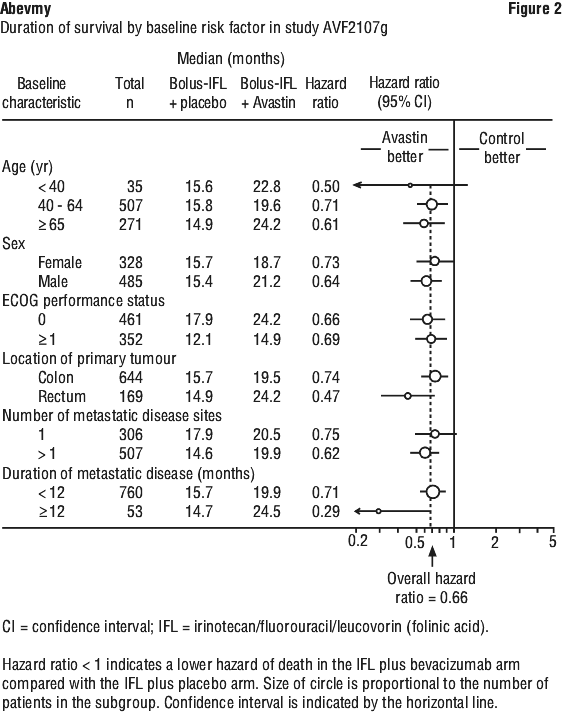
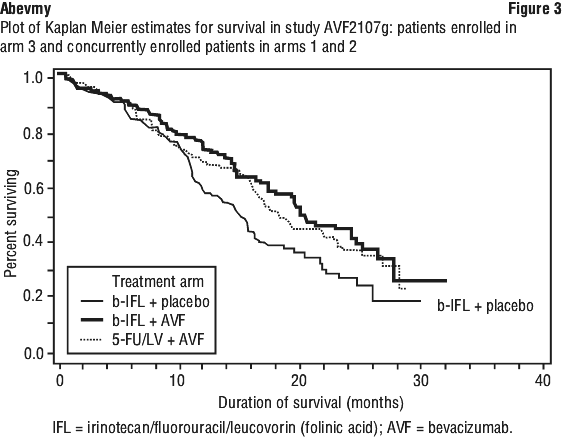
Study AVF2107g. This was a phase III randomised, double-blind, active-controlled clinical trial evaluating bevacizumab in combination with IFL as first-line treatment for metastatic colorectal cancer. Eight hundred and thirteen patients were randomised to receive IFL plus placebo (Arm 1) or IFL plus bevacizumab (Arm 2), see Table 3. A third group of 110 patients received FU/LV plus bevacizumab (Arm 3). Enrolment in Arm 3 was discontinued, as pre-specified, once safety of bevacizumab with the IFL regimen was established and considered acceptable. The median age of patients was 60 years (range 21-88), 60% were male.
Approximately 350 patients were randomised into each of the four study arms in Part II of the trial.
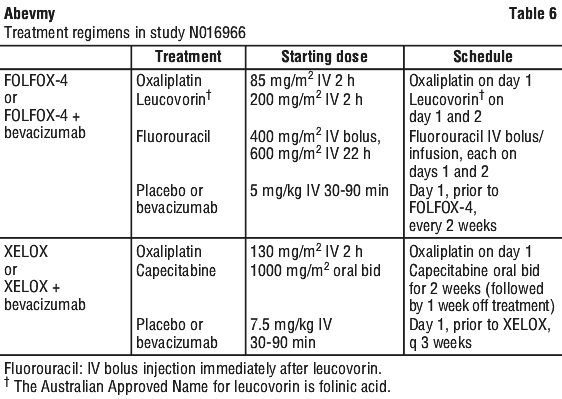
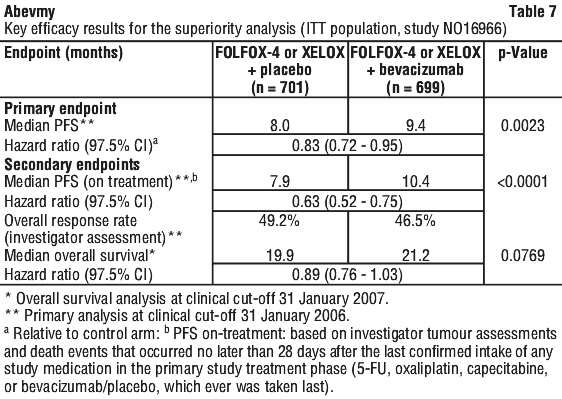
Superiority of the bevacizumab containing arms versus the chemotherapy alone arms in the overall comparison was demonstrated in terms of progression-free survival in the ITT population (see Table 7).
Secondary PFS analyses, based on Independent Review Committee and 'on-treatment'-based response assessments, confirmed the significantly superior clinical benefit for patients treated with bevacizumab.
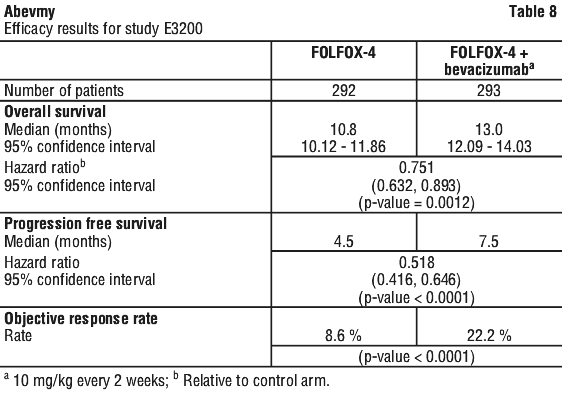
Study ECOG E3200. This was a phase III randomised, active-controlled, open-label study investigating bevacizumab 10 mg/kg in combination with leucovorin with fluorouracil bolus and then fluorouracil in fusional, with IV oxaliplatin (FOLFOX-4), administered on a 2 weekly schedule in previously treated patients (second line) with advanced colorectal cancer. In the chemotherapy arms, the FOLFOX-4 regimen used the same doses and schedule as shown in Table 6 for Study NO16966.
The primary efficacy parameter of the trial was overall survival, defined as the time from randomisation to death from any cause. Eight hundred and twenty-nine patients were randomised (292 FOLFOX-4, 293 bevacizumab+ FOLFOX-4 and 244 bevacizumab monotherapy). The addition of bevacizumab to FOLFOX-4 resulted in a statistically significant prolongation of survival. Statistically significant improvements in progression-free survival and objective response rate were also observed (see Table 8).
Adjuvant colon cancer. Study BO17920. This was a phase III randomised open-label, 3-arm study evaluating the efficacy and safety of bevacizumab administered at a dose equivalent to 2.5 mg/kg/week either every two weeks in combination with FOLFOX4, or every three weeks schedule in combination with XELOX versus FOLFOX-4 alone as adjuvant chemotherapy in 3451 patients with high-risk stage II and stage III colon carcinoma.
More relapses and deaths due to disease progression were observed in both bevacizumab arms compared to the control arm. The primary objective of prolonging disease-free survival (DFS) in patients with stage III colon cancer (n=2867) by adding bevacizumab to either chemotherapy regimen was not met. The hazard ratios for DFS were 1.17 (95% CI: 0.98-1.39) for the FOLFOX-4 + bevacizumab arm and 1.07 (95% CI: 0.90-1.28) for the XELOX + bevacizumab arm.
At the time of an exploratory interim analysis of overall survival in patients with stage III disease, 12.0% of FOLFOX-4 (control arm) patients and 15.2-15.7% of patients in the two bevacizumab-containing arms had died.
Bevacizumab is not indicated for adjuvant treatment of colon cancer.
Locally recurrent or metastatic breast cancer. (Note that the efficacy and safety of the combination of bevacizumab and paclitaxel have not been compared with anthracycline-based therapies for first-line therapy in metastatic breast cancer. The efficacy of the combination of bevacizumab and paclitaxel in second and third line treatment of metastatic breast cancer has not been demonstrated.)
E2100 was an open-label, randomised, active controlled, multicentre clinical trial evaluating bevacizumab in combination with paclitaxel for locally recurrent or metastatic breast cancer in patients who had not previously received chemotherapy for locally recurrent and metastatic disease. Prior hormonal therapy for the treatment of metastatic disease was allowed. Adjuvant taxane therapy was allowed only if it was completed at least 12 months prior to study entry.
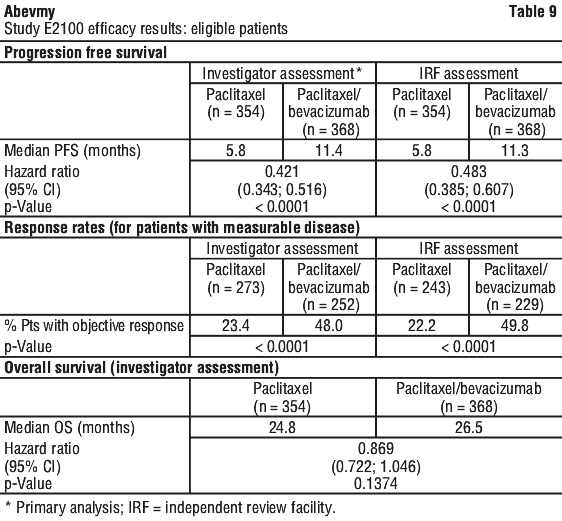
Patients were randomised to paclitaxel alone (90 mg/m2 IV over 1 hour once weekly for three out of four weeks) or in combination with bevacizumab (10 mg/kg IV infusion every two weeks). Patients were to continue assigned study treatment until disease progression. In cases where patients discontinued chemotherapy prematurely, treatment with bevacizumab as a single agent was continued until disease progression. The primary endpoint was progression free survival (PFS), as assessed by investigators. In addition, an independent review of the primary endpoint was also conducted.
Of the 722 patients in the study, the majority of patients (90%) had HER2-negative disease. A small number of patients had HER-2 receptor status that was either unknown (8%) or positive (2%). Patients who were HER2-positive had either received previous treatment with trastuzumab or were considered unsuitable for trastuzumab. The majority (65%) of patients had received adjuvant chemotherapy including 19% who had prior taxanes and 49% who had prior anthracyclines. The patient characteristics were similar between the study arms.
The results of this study are presented in Table 9 and Figure 4. The addition of bevacizumab to paclitaxel chemotherapy resulted in a significant reduction of risk of disease progression or death, as measured by PFS (HR=0.42; p < 0.0001). The resulting median PFS in bevacizumab-containing arm was 11.4 months compared with 5.8 months in the control arm. The small improvement in overall survival was not statistically significant.
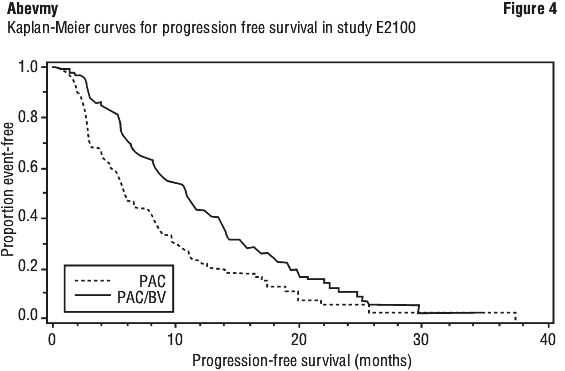
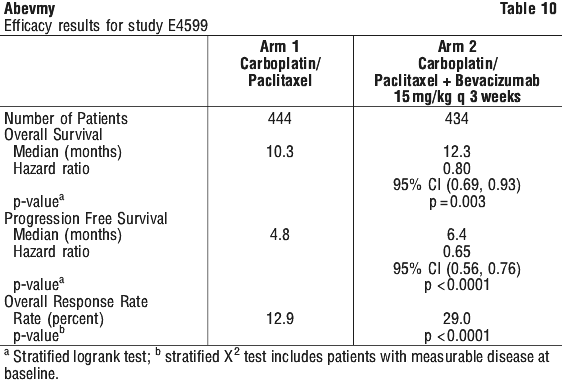
Advanced, metastatic or recurrent non-small cell lung cancer. The safety and efficacy of bevacizumab in the first-line treatment of patients with non-small cell lung cancer (NSCLC) other than predominantly squamous cell histology, was studied in addition to carboplatin/paclitaxel-based chemotherapy in study E4599 (n=878). E4599 was an open-label, randomised, active-controlled, multicentre clinical trial evaluating bevacizumab as first-line treatment of patients with locally advanced (Stage IIIB with malignant pleural effusion), metastatic or recurrent NSCLC other than predominantly squamous cell histology.
Patients were randomised to platinum-based chemotherapy (paclitaxel 200 mg/m2 and carboplatin AUC=6.0, both by IV infusion) (PC) on day 1 of every 3 week cycle for up to 6 cycles or PC in combination with bevacizumab at a dose of 15 mg/kg IV infusion day 1 of every 3 week cycle. Patients with predominant squamous histology (mixed cell type tumours only), central nervous system (CNS) metastasis, gross haemoptysis (≥ ½ tsp of red blood), clinically significant cardiovascular disease and medically uncontrolled hypertension were excluded. Other exclusion criteria were: therapeutic anticoagulation, regular use of aspirin (> 325 mg/day, NSAIDs or other agents known to inhibit platelet function, radiation therapy within 21 days of enrolment and major surgery within 28 days before enrolment.
Among 878 patients randomised to the two arms, the median age was 63, 46% were female, 43% were ≥ age 65, and 28% had ≥ 5% weight loss at study entry. 11% had recurrent disease and of the remaining 89% with newly diagnosed NSCLC, 12% had Stage IIIB with malignant pleural effusion and 76% had Stage IV disease. After completion of six cycles of carboplatin-paclitaxel chemotherapy or upon premature discontinuation of chemotherapy, patients on the bevacizumab+carboplatin-paclitaxel arm continued to receive bevacizumab as a single agent every 3 weeks until disease progression.
During the study, of the patients who received trial treatment, 32.2% (136/422) of patients received 7-12 administrations of bevacizumab and 21.1% (89/422) of patients received 13 or more administrations of bevacizumab. The primary endpoint was overall survival (OS). The secondary endpoints, PFS (progression free survival) and ORR (overall response rate), were based on investigator assessment and were not independently verified.
Overall survival was statistically significantly higher for patients receiving bevacizumab+PC chemotherapy compared with those receiving PC alone. Results are presented in Table 10.
The primary endpoint was overall survival, with secondary endpoints for the study including progression free survival (PFS). The addition of bevacizumab to IFN alfa-2a significantly increased PFS and objective tumour response rate. These results have been confirmed through an independent radiological review. However, the increase in the primary endpoint of overall survival by 2 months was not significant (HR=0.91). A high proportion of patients (approximately 63% IFN/placebo; 55% bevacizumab/IFN) received a variety of non-specified post-protocol anti-cancer therapies, including anti-neoplastic agents, which may have impacted the analysis of overall survival. The efficacy results are presented in Table 11.

Grade IV glioma. Study AVF3708g. The efficacy and safety of bevacizumab as treatment for patients with glioblastoma (GBM) was studied in an open-label, multicentre, randomised, non-comparative study (AVF3708g).
Patients in first or second relapse after prior radiotherapy (completed at least 8 weeks prior to receiving bevacizumab) and temozolomide, were randomised (1:1) to receive bevacizumab (10 mg/kg IV infusion every 2 weeks) or bevacizumab plus irinotecan (125 mg/m2 IV or 340 mg/m2 IV for patients on enzyme-inducing anti-epileptic drugs every 2 weeks) until disease progression or until unacceptable toxicity. The primary endpoints of the study were 6-month progression-free survival (PFS) and objective response rate (ORR) as assessed by an independent review facility. Other outcome measures were duration of PFS, duration of response and overall survival. Results are summarised in Table 12.
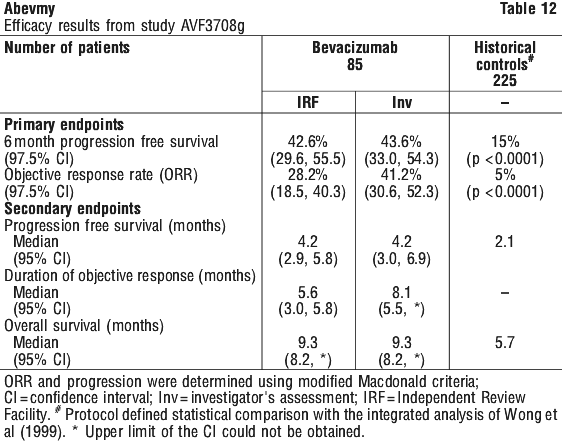
Epithelial ovarian, fallopian tube and primary peritoneal cancer. First-line ovarian cancer. Study GOG-0218. The GOG-0218 trial was a phase III multicentre, randomised, double-blind, placebo controlled, three arm study evaluating the effect of adding bevacizumab to an approved chemotherapy regimen (carboplatin and paclitaxel) in patients with optimally or sub-optimally debulked Stage III or Stage IV epithelial ovarian, fallopian tube or primary peritoneal cancer. Patients had a gynaecologic oncology performance status of 0-2 at baseline.
A total of 1873 patients were randomised in equal proportions to the following three arms:
Carboplatin/paclitaxel/placebo (CPP) arm: Placebo in combination with carboplatin (AUC 6) and paclitaxel (175 mg/m2) for 6 cycles followed by placebo alone, for a total of 15 months of therapy.
Carboplatin/paclitaxel/bevacizumab (CPB15) arm: Five cycles of bevacizumab (15 mg/kg q3w) in combination with carboplatin (AUC 6) and paclitaxel (175 mg/m2) for 6 cycles (bevacizumab commenced at cycle 2 of chemotherapy) followed by placebo alone, for a total of 15 months of therapy.
Carboplatin/paclitaxel/bevacizumab (CPB15+) arm: Five cycles of bevacizumab (15 mg/kg q3w) in combination with carboplatin (AUC 6) and paclitaxel (175 mg/m2) for 6 cycles (bevacizumab commenced at cycle 2 of chemotherapy) followed by continued use of bevacizumab (15 mg/kg q3w) as single agent for a total of 15 months of therapy.
The primary endpoint was progression-free survival (PFS) based on investigator's assessment of radiological scans. In addition, an independent review of the primary endpoint was also conducted.
The results of this study are summarised in Table 13 (the p-value boundary for primary treatment comparisons was 0.0116).
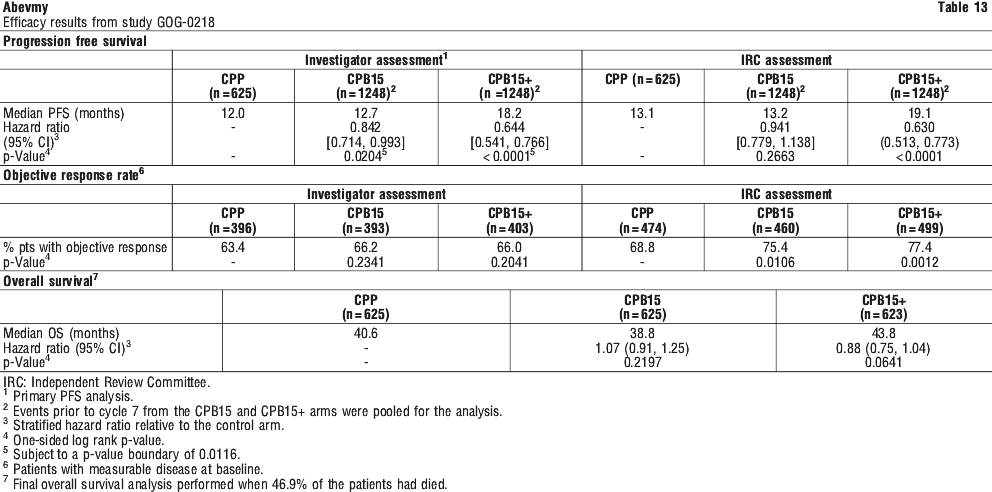
Although there was an improvement in PFS for patients who received first-line bevacizumab in combination with chemotherapy and did not continue to receive bevacizumab alone, the improvement was not statistically significant compared with patients who received chemotherapy alone.
The incidence of patients with any Grade 5 adverse event (AE) was higher in patients in the bevacizumab treated arms (2.5% CPB15+ arm and 1.6% in the CPB15 arm vs. 0.7% in the CPP arm).
Recurrent ovarian cancer. GOG-0213. GOG-0213 was a phase III randomised, controlled trial studying the safety and efficacy of bevacizumab in the treatment of patients with platinum-sensitive, recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer, who have not received prior chemotherapy in the recurrent setting. There was no exclusion criterion for prior anti-angiogenic therapy. The study evaluated the effect of adding bevacizumab to carboplatin + paclitaxel and continuing bevacizumab as a single agent compared to carboplatin + paclitaxel alone.
A total of 673 patients were randomised in equal proportions to the following two treatment arms.
CP arm: Carboplatin (AUC 5) and paclitaxel (175 mg/m2 IV over 3 hours) every 3 weeks for 6 and up to 8 cycles.
CPB arm: Carboplatin (AUC 5) and paclitaxel (175 mg/m2 IV over 3 hours) and
concurrent bevacizumab (15 mg/kg) every 3 weeks for 6 and up to 8 cycles followed by bevacizumab (15 mg/kg every 3 weeks) alone until disease progression or unacceptable toxicity.
The primary efficacy endpoint was overall survival (OS). The main secondary efficacy endpoint was progression-free survival (PFS). Objective response rates (ORR) were also examined. Results are presented in Table 14.
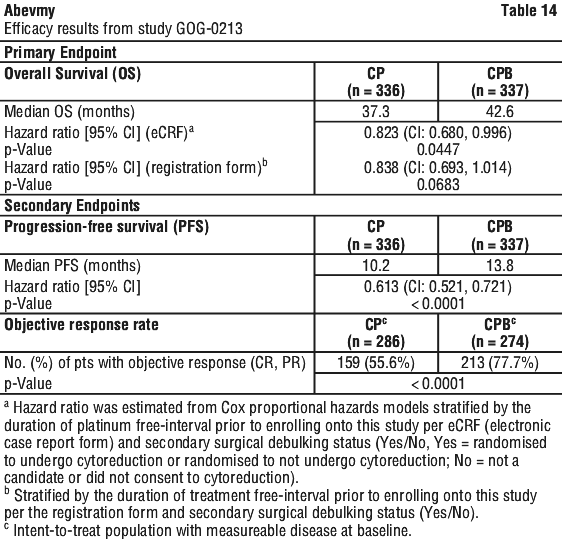
Study AVF4095g (OCEANS). The safety and efficacy of bevacizumab as treatment for patients with platinum-sensitive (defined as greater than 6 months following previous platinum therapy), recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer, who have not received prior chemotherapy in the recurrent setting, or prior bevacizumab treatment or other VEGF-targeted angiogenesis inhibitors, were studied in a phase III randomized, double blind, placebo-controlled trial (AVF4095g). The study compared the effect of adding bevacizumab to a carboplatin and gemcitabine chemotherapy followed by bevacizumab as a single agent to progression versus carboplatin and gemcitabine alone.
A total of 484 patients with measurable disease were randomized to either:
Carboplatin (AUC4 mg/mL/min, Day 1) and gemcitabine (1000 mg/m2 on Days 1 and 8) and concurrent placebo until disease progression or unacceptable toxicity.
Carboplatin (AUC4 mg/mL/min, Day 1) and gemcitabine (1000 mg/m2 on Days 1 and 8) and concurrent bevacizumab (15 mg/kg every 3 weeks) until disease progression or unacceptable toxicity.
At randomisation, patients were stratified by platinum free interval (PFI) (recurrence 6-12 months from last platinum-based treatment vs recurrence > 12 months from last platinum-based treatment) and whether they had undergone cytoreductive surgery for recurrent disease.
The primary endpoint was progression-free survival (PFS) based on investigator assessment using RECIST criteria. Additional endpoints included objective response, duration of response, safety and overall survival. An independent review of the primary endpoint was also conducted. The results of this study are summarized in Table 15.
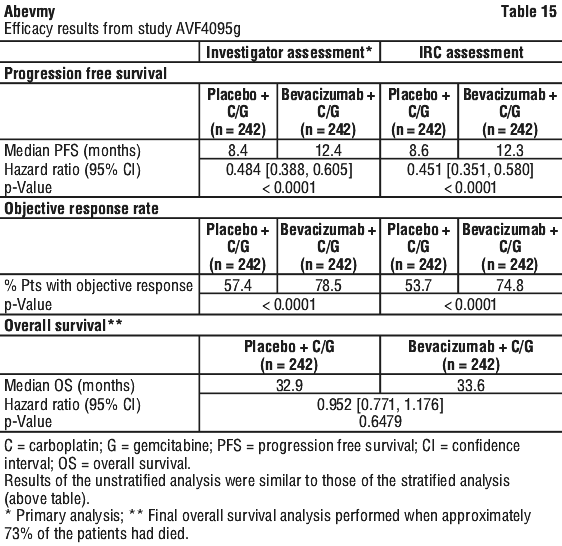
A total of 361 patients were enrolled in this study and administered either chemotherapy (paclitaxel, topotecan, or pegylated liposomal doxorubicin (PLD)) alone or in combination with bevacizumab:
CT arm (chemotherapy alone): Paclitaxel 80 mg/m2 as a 1-hour IV infusion on Days 1, 8, 15, and 22 every 4 weeks.
Topotecan 4 mg/m2 as a 30 minute IV infusion on Days 1, 8, and 15 every 4 weeks. Alternatively, a 1.25 mg/m2 dose could be administered over 30 minutes on Days 1-5 every 3 weeks.
PLD 40 mg/m2 as a 1 mg/min IV infusion on Day 1 only every 4 weeks. After cycle 1, the drug could be delivered as a 1 hour infusion.
CT + bevacizumab arm (chemotherapy plus bevacizumab): The chosen chemotherapy was combined with bevacizumab 10 mg/kg every 2 weeks or, bevacizumab 15 mg/kg every 3 weeks if used in combination with topotecan (1.25 mg/m2 on Days 1-5 on a every 3 weeks schedule).
Eligible patients had epithelial ovarian, fallopian tube or primary peritoneal cancer that progressed within 6 months of previous platinum therapy consisting of a minimum of 4 platinum therapy cycles. Patients had a life expectancy of ≥ 12 weeks, no prior radiotherapy to the pelvis or abdomen and an ECOG status ≤ 2. Exclusion criteria included patients whose disease was refractory to their previous platinum treatment, patients with previous treatment with > 2 prior anti-cancer regimens, and patients with history or symptoms of bowel obstruction, abdominal fistula, or evidence of bowel wall or recto-sigmoid involvement.
7.5% (n=27) of enrolled patients had received prior anti-angiogenic therapy. If a patient had been previously included in a blinded trial with an anti-angiogenic agent, the patient was enrolled in the same stratum as those patients who were known to have previously received an anti-angiogenic agent.
The primary endpoint was progression-free-survival (PFS), with secondary endpoints including objective response rate and overall survival. Results are presented in Table 16. PFS results for each chemotherapy cohort by Investigator and IRC assessment are presented in Table 17.
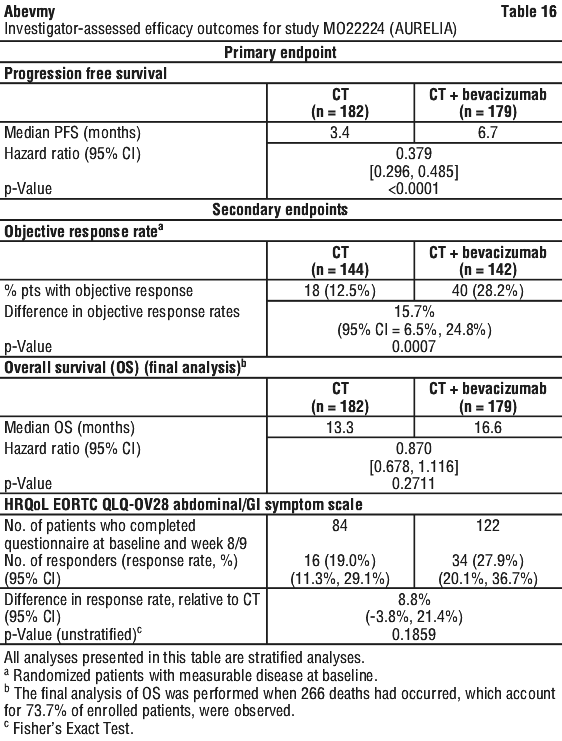
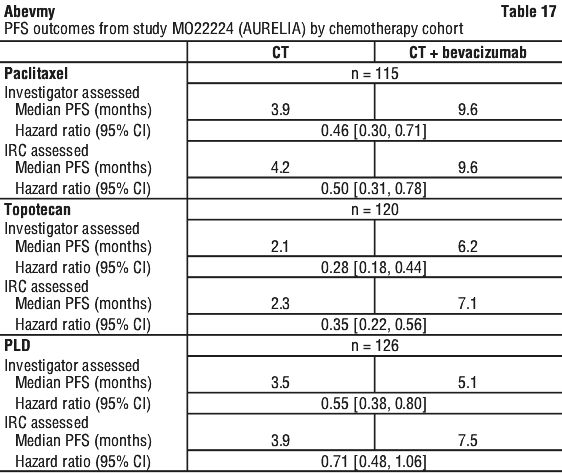
Cervical cancer. Study GOG-0240. The efficacy and safety of bevacizumab in combination with chemotherapy (paclitaxel and cisplatin or paclitaxel and topotecan) as a treatment for patients with persistent, recurrent, or Stage IVB carcinoma of the cervix (excluding patients with craniospinal metastases) was evaluated in study GOG-0240, a randomised, four-arm, multi-centre phase III trial.
A total of 452 patients were randomised to receive either:
Paclitaxel 135 mg/m2 IV over 24 hours on Day 1 and cisplatin 50 mg/m2 IV on Day 2, every 3 weeks (q3w); or paclitaxel 175 mg/m2 IV over 3 hours on Day 1 and cisplatin 50 mg/m2 IV on Day 2 (q3w); or paclitaxel 175 mg/m2 IV over 3 hours on Day 1 and cisplatin 50 mg/m2 IV on Day 1 (q3w).
Paclitaxel 135 mg/m2 IV over 24 hours on Day 1 and cisplatin 50 mg/m2 IV on Day 2 plus bevacizumab 15 mg/kg IV on Day 2 (q3w); or paclitaxel 175 mg/m2 IV over 3 hours on Day 1 and cisplatin 50 mg/m2 IV on Day 2 plus bevacizumab 15 mg/kg IV on Day 2 (q3w); or paclitaxel 175 mg/m2 IV over 3 hours on Day 1 and cisplatin 50 mg/m2 IV on Day 1 and bevacizumab 15 mg/kg IV on Day 1 (q3w).
Paclitaxel 175 mg/m2 IV over 3 hours on Day 1 and topotecan 0.75 mg/m2 IV over 30 minutes on days 1-3 (q3w).
Paclitaxel 175 mg/m2 IV over 3 hours on Day 1 and topotecan 0.75 mg/m2 IV over 30 minutes on Days 1-3 plus bevacizumab 15 mg/kg IV on Day 1 (q3w).
Eligible patients had persistent, recurrent or Stage IVB squamous cell carcinoma, adenosquamous carcinoma, or adenocarcinoma of the cervix which was not amenable to curative treatment with surgery and/or radiation therapy.
The primary efficacy endpoint was overall survival (OS). Secondary efficacy endpoints included progression-free survival (PFS) and objective response rate (ORR). Results are presented in Table 18.
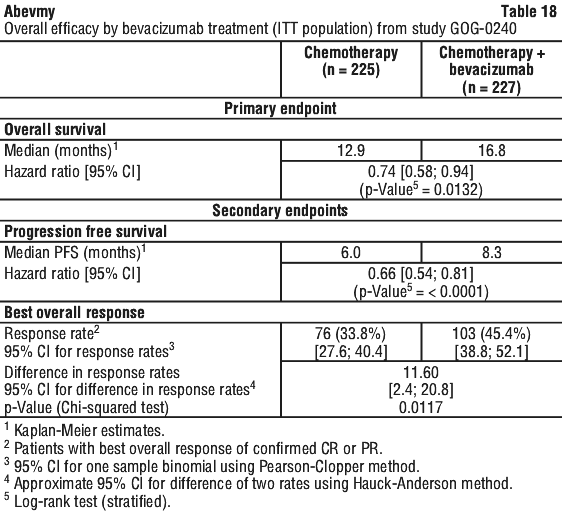
5.2 Pharmacokinetic Properties
The pharmacokinetics of bevacizumab were characterised in patients with various types of solid tumours. The doses tested were 0.1-10 mg/kg weekly in phase I; 3-20 mg/kg every two weeks (q2w) or every three weeks (q3w) in phase II; 5 mg/kg (q2w) or 15 mg/kg q3w in phase III. In all clinical trials, bevacizumab was administered as an IV infusion.
As observed with other antibodies, the pharmacokinetics of bevacizumab are well described by a two-compartment model. Overall, in all clinical trials, bevacizumab disposition was characterised by a low clearance, a limited volume of the central compartment (Vc), and a long elimination half-life. This enables target therapeutic bevacizumab plasma levels to be maintained with a range of administration schedules (such as one administration every 2 or 3 weeks).
In the population pharmacokinetics analysis, there was no significant difference in the pharmacokinetics of bevacizumab in relation to age (no correlation between bevacizumab clearance and patient age [the median age was 59 years with 5th and 95th percentiles of 37 and 76 years]).
Low albumin and high tumour burden are generally indicative of disease severity. Bevacizumab clearance was approximately 30% faster in patients with low levels of serum albumin and 7% faster in subjects with higher tumour burden when compared with the typical patient with median values of albumin and tumour burden.
Absorption. Not applicable.
Distribution. The typical value for central volume (Vc) was 2.73 L and 3.28 L for female and male patients, respectively, which is in the range that has been described for IgGs and other monoclonal antibodies. After correcting for body weight, male patients had a larger Vc (+20%) than female patients.
Metabolism. Assessment of bevacizumab metabolism in rabbits following a single IV dose of 125I-bevacizumab suggested that its metabolic profile was similar to that expected for a native IgG molecule which does not bind VEGF.
Excretion. The pharmacokinetics of bevacizumab are linear at doses ranging from 1.5 to 10 mg/kg/wk.
The value for clearance is, on average, equal to 0.188 and 0.220 L/day for female and male patients, respectively. After correcting for body weight, male patients had a higher bevacizumab clearance (+17%) than females. According to the two-compartmental model, the elimination half-life is 18 days for a typical female patient and 20 days for a typical male patient.
Pharmacokinetics in special populations. The population pharmacokinetics of bevacizumab were analysed to evaluate the effects of demographic characteristics. In adults, the results showed no significant difference in the pharmacokinetics of bevacizumab in relation to age.
Children and adolescents. The pharmacokinetics of bevacizumab were evaluated in 152 patients (7 months to 21 years; 5.9 to 125 kg) across 4 clinical studies using a population pharmacokinetic model. The pharmacokinetic results show that the clearance and the volume of distribution of bevacizumab were comparable between paediatric and adult patients when normalised by body-weight. Age was not associated with the pharmacokinetics of bevacizumab when bodyweight was taken into account.
Renal impairment. No studies have been conducted to investigate the pharmacokinetics of bevacizumab in renally impaired patients since the kidneys are not a major organ for bevacizumab metabolism or excretion.
Hepatic impairment. No studies have been conducted to investigate the pharmacokinetics of bevacizumab in patients with hepatic impairment since the liver is not a major organ for bevacizumab metabolism or excretion.
Patients with ascites. No studies have examined the effect of ascites on the pharmacokinetic parameters of bevacizumab.
5.3 Preclinical Safety Data
Genotoxicity. No data available.
Carcinogenicity. Studies to evaluate the carcinogenic and mutagenic potential of bevacizumab have not been performed.
6 Pharmaceutical Particulars
6.1 List of Excipients
Abevmy also contains trehalose dihydrate, monobasic sodium phosphate dihydrate, dibasic sodium phosphate, polysorbate 20, water for injections, sodium hydroxide and ortho phosphoric acid.
6.2 Incompatibilities
Incompatibilities were not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store vials at 2-8°C. (Refrigerate. Do not freeze.). Keep vial in carton to protect from light.
Abevmy does not contain any antimicrobial agent; therefore, care must be taken to ensure the sterility of the prepared solution. Product is for single use in one patient only. Discard any residue. Parenteral drug products should be inspected visually for particulate matter and discolouration prior to administration.
Chemical and physical in-use stability has been demonstrated for 48 hours at 2°C to 30°C in 0.9% sodium chloride solution.
Abevmy is microbiologically stable for 24 hours at 2°C to 30°C in PVC and PO bags containing 0.9% sodium chloride solution. To reduce microbiological hazard, the product should be used as soon as practicable after preparation. If storage is necessary, do not store longer than 24 hours at 2-8°C.
6.5 Nature and Contents of Container
Container type. Clear Type I glass, with a latex free chlorobutyl rubber stopper and aluminium flip off seal.
Pack sizes. 100 mg pack containing one 4 mL single-dose vial.
400 mg pack containing one 16 mL single-dose vial.
Australian register of therapeutic goods (ARTG). AUST R 334816 - Abevmy bevacizumab 100 mg/4 mL concentrate injection for solution vial.
AUST R 334814 - Abevmy bevacizumab 400 mg/16 mL concentrate injection for solution vial.
6.6 Special Precautions for Disposal
The release of medicines into the environment should be minimised. Medicines should not be disposed of via wastewater and disposal through household waste should be avoided. Any unused medicine or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
Chemical structure. Bevacizumab is an immunoglobulin G (IgG) composed of two identical light chains, consisting of 214 amino acid residues and two 453 residue heavy chains containing an N-linked oligosaccharide and has a molecular weight of approximately 149,000 daltons.
CAS number. 216974-75-3.
7 Medicine Schedule (Poisons Standard)
S4 (Prescription Only Medicine).
Date of First Approval
06 September 2021
Date of Revision
11 March 2025
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.