Adcetris
Brand Information
| Brand name | Adcetris |
| Active ingredient | Brentuximab vedotin |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Adcetris.
Summary CMI
Adcetris®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I using Adcetris?
Adcetris contains the active ingredient brentuximab vedotin. Adcetris is used to treat patients with Hodgkin Lymphoma who have not had treatment before or to treat Hodgkin Lymphoma that has come back or not responded to previous treatment. Adcetris may be used to lower the likelihood of Hodgkin Lymphoma coming back after a stem cell transplant in patients with certain risk factors. Adcetris is also used to treat patients with cutaneous T-Cell Lymphoma who have previously received at least one anti-cancer medicine that travels through the bloodstream and to treat patients with Peripheral T-Cell Lymphoma who have not had treatment before. Adcetris may also be used to treat Systemic Anaplastic Large Cell Lymphoma that has come back or not responded to previous treatment. For more information, see Section 1. Why am I using Adcetris?.
2. What should I know before I use Adcetris?
Do not use if you have ever had an allergic reaction to brentuximab vedotin or any of the ingredients listed at the end of the CMI. Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding. For more information, see Section 2. What should I know before I use Adcetris? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with Adcetris and affect how it works. A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use Adcetris?
Adcetris will be given to you by your doctor or nurse, through a drip in one of the veins in your arm (intravenous infusion) over about 30 minutes. More instructions can be found in Section 4. How do I use Adcetris? in the full CMI.
5. What should I know while using Adcetris?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using Adcetris? in the full CMI.
6. Are there any side effects?
Upper respiratory tract infection, pneumonia, back pain, constipation, nausea, vomiting, feeling tired, frequent urination, increased thirst, change in appetite, weight loss, irritability, unusual bleeding/bruising under skin or bleeding gums, headaches, dizziness, looking pale, itching, hair loss/thinning, muscle pain, joint pain or painful, swollen joints, blisters which may crust/scab, skin redness, pain, swelling, blistering or peeling at infusion site, increased liver enzymes, new or recurring cytomegalovirus infection, sore creamy-yellow raised patches in the mouth, cold sores, trouble sleeping, pain on urination. Serious side effects: confusion or trouble thinking clearly, memory loss, blurred vision, loss of vision, decreased strength/control or sensation in arms or legs, change in way you walk, balance problems, new or worsening shortness of breath or cough, decreased urination, heart rhythm disturbances, change sensitivity of skin, numbness, tingling, discomfort, burning sensation or weakness or pain in hands or feet, weakness and difficulty walking, loss of appetite, pain in upper right side of stomach, yellowing of skin or white part of eyes, severe skin problems such as itchiness, redness, rash with swelling, blistering or peeling or rash when exposed to sun, possibly with joint pain and general fever, swelling of face, lips, mouth tongue or throat which may cause difficulty swallowing or breathing, severe upper stomach pain with or without nausea and vomiting or stomach pain that spreads to your back.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
Adcetris®
Active ingredient(s): brentuximab vedotin
Consumer Medicine Information (CMI)
This leaflet provides important information about using Adcetris. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using Adcetris.
Where to find information in this leaflet:
1. Why am I using Adcetris
2. What should I know before I use Adcetris?
3. What if I am taking other medicines?
4. How do I use Adcetris?
5. What should I know while using Adcetris?
6. Are there any side effects?
7. Product details
1. Why am I using Adcetris
Adcetris contains the active ingredient brentuximab vedotin.
Adcetris belongs to a group of medicines known as anti-cancer agents. There are many different classes of anti-cancer agents.
Adcetris is designed to work differently than traditional anti-cancer agents (chemotherapy). Traditional chemotherapy enters the blood and kills both cancer cells and healthy cells that divide rapidly. Adcetris is made up of a monoclonal antibody linked to a substance intended to kill cancer cells. This substance is delivered to cancer cells by the monoclonal antibody.
A monoclonal antibody is a protein which recognises certain cancer cells.
Hodgkin Lymphoma
Adcetris is used to treat patients with Hodgkin Lymphoma who have not had treatment before. When Adcetris is used to treat Hodgkin Lymphoma that has not already been treated, it is given in combination with other chemotherapy medicines used to treat this condition.
Adcetris is also used to treat Hodgkin Lymphoma that has come back or not responded to previous treatment.
Adcetris may also be used alone to lower the likelihood of Hodgkin Lymphoma coming back after a stem cell transplant in patients with certain risk factors. In these patients, Adcetris may help prevent or delay recurrence of disease. Your doctor will discuss the potential risks and benefits of receiving Adcetris following a stem cell transplant.
Hodgkin Lymphoma is a type of cancer of the white blood cells.
Cutaneous T-Cell Lymphoma
Adcetris is also used to treat patients with cutaneous T-Cell Lymphoma (CTCL) who have previously received at least one anti-cancer medicine that travels through the bloodstream.
CTCL is a cancer of a certain type of white blood cell called a ‘T-cell’ that mainly affects the skin. Adcetris is used to treat CTCL where a specific type of protein is present on the cells' surface.
Peripheral T-Cell Lymphoma
Adcetris is also used to treat patients with Peripheral T-Cell Lymphoma (PTCL) who have not had treatment before.
PTCL is a type of non Hodgkin Lymphoma found in the lymph nodes and/or throughout other parts of the body. When Adcetris is used to treat PTCL that has not already been treated, it is given in combination with other chemotherapy medicines used to treat this condition.
Systemic Anaplastic Large Cell Lymphoma (sALCL) is a type of PTCL. Adcetris may also be used alone to treat sALCL that has come back or not responded to previous treatment.
2. What should I know before I use Adcetris?
Warnings
Do not use Adcetris if:
- you are allergic to brentuximab vedotin, or any of the ingredients listed at the end of this leaflet.
- some symptoms of an allergic reaction include skin rash, itching, shortness of breath or swelling of the face, lips or tongue, which may cause difficulty in swallowing or breathing.
- you are currently taking a medicine called bleomycin, an anti-cancer agent.
- always check the ingredients to make sure you can use this medicine.
- the expiry date printed on the pack has passed or if the packaging is damaged or shows signs of tampering. If it has expired or is damaged return it to your pharmacist for disposal.
Check with your doctor if you:
- have any allergies to any other medicines, foods, preservatives or dyes.
- have, or think you have, an infection.
- are taking, or have previously taken, medicines which may affect your immune system, such as chemotherapy or immunosuppressive agents. If you are taking or have taken medicines which affect your immune system, you may have an increased risk of infections.
- have or have had any problems with your kidneys or liver.
- have any other medical conditions.
- take any medicines for any other condition
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Tell your doctor if you are intending to get pregnant or father a child. You and your partner must use two methods of effective contraception during your treatment with this medicine. Women must continue using contraception for 6 months following the last dose of Adcetris.
Men being treated with Adcetris are advised to have sperm samples frozen and stored before treatment. Men are advised not to father a child during treatment with this medicine and for up to 6 months following the last dose of this medicine.
Tell your doctor if you are pregnant, think you may be pregnant or are breastfeeding. Your doctor can discuss the risks and benefits involved.
Use in Children
Adcetris is to be given to adults only. There is not enough information to recommend the use of this medicine for children under the age of 18 years.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interfere with Adcetris and affect how it works. These include:
- medicines used in the treatment of fungal infections such as ketoconazole or itraconazole.
These medicines may be affected by Adcetris, or may affect how well it works. You may need to use different amounts of your medicine, or take different medicines.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect Adcetris.
4. How do I use Adcetris?
How much to use
- Your doctor will decide what dose and how long you will receive Adcetris.
- The dose of Adcetris depends on your body weight.
- The usual recommended dose of Adcetris when it is given alone is 1.8 mg/kg, given once every 3 weeks. Your doctor may lower your starting dose if you have kidney or liver problems.
- If you are a patient with Hodgkin Lymphoma that has not already been treated, you will receive Adcetris in combination with other chemotherapy medicines. You may receive Adcetris in combination with doxorubicin, vinblastine and dacarbazine, or, you may receive Adcetris in combination with etoposide, cyclophosphamide, doxorubicin, dacarbazine, and dexamethasone. Your doctor will decide which combination is right for you.
See the Consumer Medicine Information leaflets for these medicines given in combination with Adcetris for additional information on their use and effects.
The usual dose of Adcetris given in combination with doxorubicin, vinblastine and dacarbazine is 1.2 mg/kg given every 2 weeks for 6 months. Your doctor may lower your starting dose if you have mild liver problems.
The usual dose of Adcetris given in combination with etoposide, cyclophosphamide, doxorubicin, dacarbazine, and dexamethasone is 1.8 mg/kg given every 3 weeks for up to 6 months. - If you are a patient with PTCL that has not already been treated, you will receive Adcetris in combination with cyclophosphamide, doxorubicin, and prednisone which are other medicines used to treat this condition.
See the Consumer Medicine Information leaflets for these medicines given in combination with Adcetris for additional information on their use and effects.
The usual dose of Adcetris given in combination with cyclophosphamide, doxorubicin, and prednisone is 1.8 mg/kg given every 3 weeks for approximately 4 - 6 months. Your doctor may lower your starting dose if you have mild liver problems. - After the first dose of Adcetris in combination with chemotherapy, your doctor may also give you a medicine that will help prevent development or reduce the severity of neutropenia (decrease of white blood cell count) which can increase the risk of infection.
How Adcetris is given
- Adcetris will be given to you by your doctor or nurse, through a drip in one of the veins in your arm (intravenous infusion) over about 30 minutes.
- Your healthcare provider will monitor you during and after the Adcetris infusion for side effects to see if you have a reaction to the treatment.
How long to take it
- Continue receiving your medicine for as long as your doctor tells you.
You should not stop using Adcetris without talking with your doctor first.
If you forget to use Adcetris
If you forget or miss an appointment to receive the infusion, make another appointment as soon as possible.
If you are not sure what to do, ask your doctor.
If you use too much Adcetris
As Adcetris is given to you under the supervision of your doctor, it is very unlikely that you will receive too much.
However, if you experience any side effects after being given Adcetris, tell your doctor or nurse immediately.
If you think that you have used too much Adcetris, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using Adcetris?
Things you must do
- If you are about to be started on any new medicine, remind your doctor and pharmacist that you are receiving Adcetris.
- Tell any other doctors, dentists and pharmacists who treat you that you are receiving this medicine.
- If you are going to have surgery, tell the surgeon that you are receiving this medicine.
- Keep all of your doctor's appointments so that your progress can be checked. Your doctor may do regular blood tests to make sure it is safe for you to receive this medicine and to help prevent unwanted side-effects. Some patients receiving Adcetris may develop some serious conditions requiring immediate treatment.
Tell your doctor straight away if you notice any of the following symptoms because some of them may be signs of a serious or possibly fatal condition:
Progressive multifocal leukoencephalopathy (PML)
PML is a serious and life-threatening brain condition.
Tell your partner or caregiver you are receiving Adcetris and ask them to tell you if they notice any changes in your movement or behaviour.
Symptoms of PML can include:
- confusion or trouble thinking clearly
- memory loss,
- blurred vision or loss of vision
- decreased strength/control or sensation in your arms or legs, a change in the way you walk or problems with your balance
Lung problems
Tell your doctor straight away if you develop new or worsening shortness of breath or cough.
These could be symptoms of a side-effect called pulmonary toxicity.
Liver injury
Tell your doctor straight away if you develop a loss of appetite, pain in the upper right side of your stomach area, nausea, vomiting, yellowing of your skin or the white part of your eyes (jaundice).
These could be symptoms of a side-effect called hepatotoxicity.
Inflammation of the pancreas
Tell your doctor straight away if you get any of the following symptoms:
- severe upper stomach pain with or without nausea and vomiting or stomach pain that spreads to your back.
These could be symptoms of a condition called pancreatitis.
Infection
Tell your doctor right away if you get any of the following symptoms:
- fever (greater than or equal to 38°C) and/or chills or shivering
- sore throat
- cough
- pain on urination
These could be symptoms of an infection and/or caused by a condition called febrile neutropenia (lack of white blood cells).
Infusion Reactions
Medicines of this type (monoclonal antibodies) can cause infusion reactions. In general, these types of reactions occur within minutes to several hours following completion of the infusion. However, they may develop more than several hours after completion of the infusion but this is uncommon. Symptoms of infusion reactions include:
- rash, shortness of breath, difficulty breathing or a tight chest, fever and back pain.
If you think you have previously had a similar reaction, tell your doctor before you are given this medicine.
Severe Skin Reactions
Tell your doctor right away if you experience flu-like symptoms followed by a painful red or purplish rash that spreads and blisters.
These could be symptoms of rare, serious disorders called Stevens-Johnson syndrome, Toxic Epidermal Necrolysis and Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS).
Gastrointestinal (bowel) problems
Tell your doctor straight away if you get any of the following symptoms:
- severe stomach pain,
- chills
- nausea, vomiting or diarrhoea
Other serious symptoms
Tell your doctor right away if you get any of the following symptoms:
- hypersensitivity reaction called DRESS (Drug Reaction with Eosinophilia and Systemic Symptoms) which may include fever, extensive skin rash, swollen lymph nodes, blood abnormalities and inflammation of internal organs like the liver, lungs or kidneys
- a potentially-life threatening condition called tumour lysis syndrome in which you may experience dizziness, decreased urination, confusion, vomiting, nausea, swelling, shortness of breath, or heart rhythm disturbances
- a condition called peripheral neuropathy which can change the sensitivity of the skin, causing symptoms such as numbness, tingling, discomfort, a burning sensation, weakness, or pain in the hands or feet
- a condition called motor neuropathy, which can cause symptoms that include a feeling of weakness and difficulty walking
The above list includes very serious side effects. You may need urgent medical attention. Many of these side effects are rare.
Things you should not do
- Do not use this medicine to treat any other complaints unless your doctor tells you to.
- Do not give this medicine to anyone else, even if they have the same condition as you.
- Do not stop using Adcetris without checking with your doctor.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how Adcetris affects you. This medicine may have a minor influence on your ability to drive or use tools or machines.
Adcetris may cause dizziness in some people.
If you feel dizzy, do not drive or use tools or machines.
Looking after your medicine
- Keep the unopened vial in a refrigerator stored between 2-8 degrees C. Do not freeze.
- Keep the vial in the original carton in order to protect from light.
- Do not use this medicine after the expiry date.
Follow the instructions in the carton on how to take care of your medicine properly.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Keep it where young children cannot reach it.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What Adcetris contains
| Active ingredient (main ingredient) | Each vial of Adcetris contains 50 mg of brentuximab vedotin. After reconstitution each mL of solution contains 5 mg of brentuximab vedotin. |
| Other ingredients (inactive ingredients) | trehalose dihydrate sodium citrate dihydrate citric acid monohydrate polysorbate 80 |
Do not take this medicine if you are allergic to any of these ingredients.
Adcetris does not contain gluten, sucrose, lactose, tartrazine or any other azo dyes.
What Adcetris looks like
Adcetris is a white to off-white cake or powder provided in a glass vial.
Each pack of Adcetris consists of one vial (AUST R 203372).
Who distributes Adcetris
Adcetris is supplied in Australia by:
Takeda Pharmaceuticals Australia Pty Ltd
Level 39, 225 George Street
Sydney NSW 2000
Ph: 1800 012 612
www.takeda.com/en-au
This leaflet was prepared in December 2025
ADCETRIS and the ADCETRIS Logo are registered trademarks of Millennium Pharmaceuticals, Inc., a Takeda Company.
TAKEDA and the TAKEDA Logo are registered trademarks of Takeda Pharmaceutical Company Limited.
Brand Information
| Brand name | Adcetris |
| Active ingredient | Brentuximab vedotin |
| Schedule | S4 |
MIMS Revision Date: 01 February 2026
1 Name of Medicine
Brentuximab vedotin.
2 Qualitative and Quantitative Composition
Adcetris contains 50 mg brentuximab vedotin per vial. Following reconstitution with 10.5 mL sterile water for injection, a solution containing 5 mg/mL brentuximab vedotin is produced.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Powder for injection. White to off-white lyophilized cake or powder.
4 Clinical Particulars
4.1 Therapeutic Indications
Hodgkin lymphoma. Treatment of patients with previously untreated CD30+ stage III or stage IV Hodgkin lymphoma (HL) in combination with doxorubicin, vinblastine, and dacarbazine (AVD).
Treatment of adult patients with previously untreated CD30+ stage IIB with large mediastinal mass and/or extranodal disease, stage III or stage IV HL in combination with etoposide, cyclophosphamide, doxorubicin, dacarbazine, dexamethasone (BrECADD).
Treatment of adult patients with CD30+ HL at higher risk of relapse or progression following ASCT.
Treatment of adult patients with relapsed or refractory CD30+ HL:
1. following autologous stem cell transplant (ASCT); or
2. following at least two prior therapies when ASCT or multi-agent chemotherapy is not a treatment option.
Peripheral T-cell lymphoma. Treatment of adult patients with previously untreated CD30+ peripheral T-cell lymphoma (PTCL) in combination with cyclophosphamide, doxorubicin, and prednisone (CHP).
Treatment of adult patients with relapsed or refractory systemic anaplastic large cell lymphoma (sALCL).
Cutaneous T-cell lymphoma. Treatment of adult patients with CD30+ cutaneous T-cell lymphoma (CTCL) after at least 1 prior systemic therapy.
4.2 Dose and Method of Administration
Adcetris should be administered under the supervision of a physician experienced in the use of anti-cancer agents.
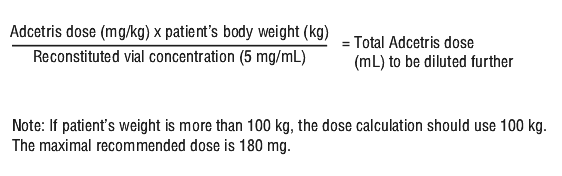
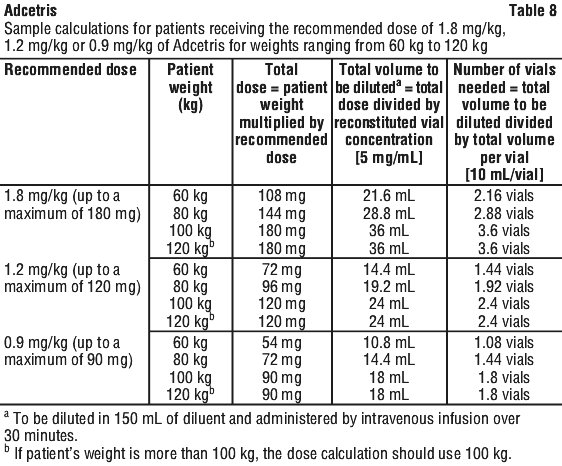
Posology. Previously untreated HL. Adcetris in combination with doxorubicin [A], vinblastine [V] and dacarbazine [D] [A+AVD]. The recommended dose in combination with chemotherapy (doxorubicin [A], vinblastine [V] and dacarbazine [D] [AVD]) is 1.2 mg/kg administered as an intravenous infusion over 30 minutes on days 1 and 15 of each 28-day cycle for 6 cycles (see Section 5.1 Pharmacodynamic Properties, Clinical trials). If the patient's weight is more than 100 kg, the dose calculation should use 100 kg (see Table 8).
Primary prophylaxis with G-CSF, beginning with the first dose, is recommended for all patients with previously untreated HL receiving combination therapy (see Section 4.4 Special Warnings and Precautions for Use).
Refer to the product information of chemotherapy agents given in combination with Adcetris for the treatment of patients with previously untreated HL.
Adcetris in combination with etoposide [E], cyclophosphamide [C], doxorubicin [A], dacarbazine [D], dexamethasone [D] [BrECADD]. The recommended dose in combination with chemotherapy (etoposide [E], cyclophosphamide [C], doxorubicin [A], dacarbazine [D], dexamethasone [D] [BrECADD]) is 1.8 mg/kg administered as an intravenous infusion over 30 minutes every 3 weeks for up to 6 cycles (see Section 5.1 Pharmacodynamic Properties, Clinical trials). If the patient's weight is more than 100 kg, the dose calculation should use 100 kg (see Table 8).
Primary prophylaxis with growth factor support (G-CSF) must be given beginning on day 5 of each cycle, for all adult patients receiving BrECADD (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Pretreatment with dexamethasone for 4 days before the first cycle of chemotherapy is recommended for patients > 40 years of age or at physician's discretion.
An antibiotic prophylaxis must be given 3 times a week during the whole duration of chemotherapy.
See Table 5 for dosing recommendations for chemotherapy agents given in combination with Adcetris for patients with previously untreated HL.
Relapsed or refractory HL. The recommended dose is 1.8 mg/kg administered as an intravenous infusion over 30 minutes every 3 weeks. If the patient's weight is more than 100 kg, the dose calculation should use 100 kg (see Table 8).
The recommended starting dose for the retreatment of patients who have previously responded to treatment with Adcetris is 1.8 mg/kg administered as an intravenous infusion over 30 minutes every 3 weeks. Alternatively, treatment may be started at the last tolerated dose.
Patients who achieve stable disease or better should receive a minimum of 8 cycles and up to a maximum of 16 cycles (approximately 1 year) (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
HL at risk of relapse or progression following ASCT. The recommended dose is 1.8 mg/kg administered as an intravenous infusion over 30 minutes every 3 weeks. If the patient's weight is more than 100 kg, the dose calculation should use 100 kg (see Table 8).
Adcetris treatment should start following recovery from ASCT based on medical judgment. These patients should receive up to 16 cycles (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Previously untreated PTCL. The recommended dose in combination with chemotherapy (cyclophosphamide [C], doxorubicin [H], and prednisone [P]; [CHP]) is 1.8 mg/kg administered as an intravenous infusion over 30 minutes every 3 weeks for 6 to 8 cycles. If the patient's weight is more than 100 kg, the dose calculation should use 100 kg (see Table 8).
Primary prophylaxis with G-CSF, beginning with the first dose, is recommended for all patients with previously untreated PTCL receiving combination therapy (see Section 4.4 Special Warnings and Precautions for Use).
Refer to the product information of chemotherapy agents given in combination with Adcetris for patients with previously untreated PTCL.
Relapsed or refractory sALCL. The recommended dose is 1.8 mg/kg administered as an intravenous infusion over 30 minutes every 3 weeks. If the patient's weight is more than 100 kg, the dose calculation should use 100 kg (see Table 8).
The recommended starting dose for the retreatment of patients who have previously responded to treatment with Adcetris is 1.8 mg/kg administered as an intravenous infusion over 30 minutes every 3 weeks. Alternatively, treatment may be started at the last tolerated dose.
Patients who achieve stable disease or better should receive a minimum of 8 cycles and up to a maximum of 16 cycles (approximately 1 year) (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Relapsed or refractory CTCL after prior systemic treatment. The recommended dose is 1.8 mg/kg administered as an intravenous infusion over 30 minutes every 3 weeks. If the patient's weight is more than 100 kg, the dose calculation should use 100 kg (see Table 8).
Patients with CTCL should receive up to 16 cycles (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
General. Complete blood counts and hepatic function tests should be monitored prior to administration of each dose of this treatment (see Section 4.4 Special Warnings and Precautions for Use).
Patients should be monitored during and after infusion (see Section 4.4 Special Warnings and Precautions for Use).
Dose adjustments. Renal and hepatic impairment. Monotherapy. Adcetris should be used with caution in patients with severe renal impairment due to the potential for increased exposure of monomethyl auristatin E (MMAE) and increased toxicity (see Section 5.2 Pharmacokinetic Properties). The following dosage is recommended:
Mild (creatinine clearance 50-80 mL/min) or moderate (creatinine clearance 30-50 mL/min) renal impairment: 1.8 mg/kg every 3 weeks;
Severe (creatinine clearance less than 30 mL/min) renal impairment: in patients with active disease and no other treatment options, consider use with caution after evaluating benefit/ risk at a starting dose of 1.2 mg/kg every 3 weeks;
Dialysis dependent: no information.
Adcetris should be used with caution in patients with hepatic impairment due to the potential for increased exposure of MMAE and increased toxicity (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties).
The following dosage is recommended:
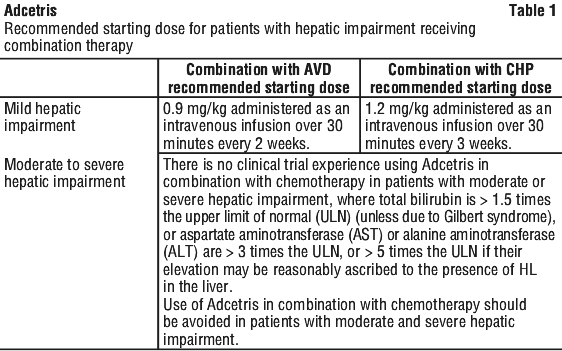
Mild (Child-Pugh A) hepatic impairment: 1.2 mg/kg every 3 weeks;
Moderate (Child-Pugh B) or severe (Child-Pugh C) hepatic impairment: in patients with active disease and no other treatment options, consider use with caution after evaluating benefit/ risk at a starting dose of 1.2 mg/kg every 3 weeks.
Patients should be closely monitored for the development of serious hepatotoxicity during Adcetris therapy as this risk may be greater with pre-existing hepatic impairment.
Combination therapy. Patients with renal impairment should be closely monitored for adverse events. There is no clinical trial experience using Adcetris in combination with chemotherapy in patients with renal impairment, where serum creatinine is ≥ 0.177 mmol/L (2.0 mg/dL) and/or creatinine clearance or calculated creatinine clearance is ≤ 40 mL/minute. Use of Adcetris in combination with chemotherapy should be avoided in patients with severe renal impairment.
Patients with hepatic impairment should be closely monitored for adverse events.
See Table 1 for starting dose recommendations for patients with hepatic impairment receiving combination therapy.


If events occur in two successive cycles, the dose should be reduced to baseline level shown in Table 5.

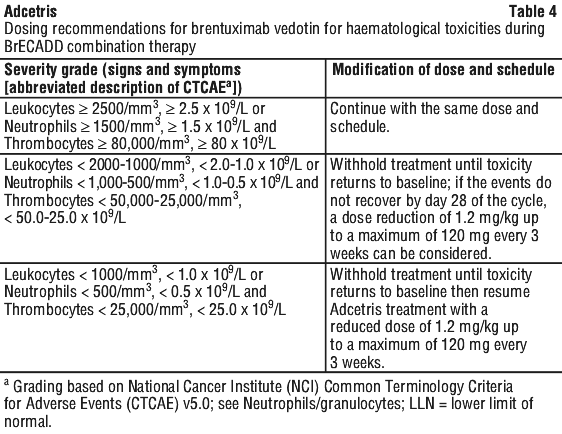
Peripheral neuropathy. If peripheral sensory or motor neuropathy emerges or worsens during treatment see Table 6 and Table 7 for appropriate dosing recommendations for monotherapy and combination therapy, respectively (see Section 4.4 Special Warnings and Precautions for Use).


Adcetris must not be administered as an intravenous push or bolus. Adcetris should be administered through a dedicated intravenous line and it must not be mixed with other medicinal products.
Procedures for proper handling and disposal of anticancer medicines should be considered.
Proper aseptic technique throughout the handling of this medicinal product should be followed.
Instructions for reconstitution. Each single use vial must be reconstituted with 10.5 mL of water for injections to a final concentration of 5 mg/mL.
1. Direct the stream toward the wall of the vial and not directly at the cake or powder.
2. Gently swirl the vial to aid dissolution. Do not shake.
3. The reconstituted solution in the vial is a clear to slightly opalescent, colourless solution with a final pH of 6.6.
4. The reconstituted solution should be inspected visually for any foreign particulate matter and/or discoloration. In the event of either being observed, discard the medicinal product.
Preparation of infusion solution. The appropriate amount of reconstituted Adcetris must be withdrawn from the vial(s) and added to an infusion bag containing sodium chloride 9 mg/mL (0.9%) solution for injection in order to achieve a final concentration of 0.4-1.2 mg/mL Adcetris. The recommended diluent volume is 150 mL. The already reconstituted Adcetris can also be diluted into 5% dextrose for injection or lactated Ringer's for injection.
Gently invert the bag to mix the solution containing Adcetris. Do not shake.
Any portion left in the vial, after withdrawal of the volume to be diluted, must be disposed of in accordance with local requirements.
Do not add other medicinal products to the prepared Adcetris infusion solution or intravenous infusion set. The infusion line should be flushed following administration with sodium chloride 9 mg/mL (0.9%) solution for injection, 5% dextrose for injection, or lactated Ringer's for injection. Following dilution, infuse the Adcetris solution immediately at the recommended infusion rate.
Total storage time of the solution from reconstitution to infusion should not exceed 24 hours.
Determining dosage amount. Calculation to determine the total Adcetris dose (mL) to be further diluted: see Equation 1.
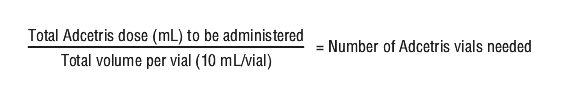
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients (see Section 6.1 List of Excipients).
Combined use of bleomycin and Adcetris causes pulmonary toxicity.
4.4 Special Warnings and Precautions for Use
Progressive multifocal leukoencephalopathy. John Cunningham virus (JCV) reactivation resulting in progressive multifocal leukoencephalopathy (PML) and death can occur in Adcetris-treated patients. PML has been reported in patients who received this treatment after receiving multiple prior chemotherapy regimens. PML is a rare demyelinating disease of the central nervous system that results from reactivation of latent JCV and is often fatal.
Patients should be closely monitored for new or worsening neurological, cognitive, or behavioural signs or symptoms, which may be suggestive of PML. Adcetris dosing should be held for any suspected case of PML. Suggested evaluation of PML includes neurology consultation, gadolinium-enhanced magnetic resonance imaging of the brain and cerebrospinal fluid analysis for JCV DNA by polymerase chain reaction or a brain biopsy with evidence of JCV. A negative JCV PCR does not exclude PML. Additional follow up and evaluation may be warranted if no alternative diagnosis can be established. Adcetris dosing should be permanently discontinued if a diagnosis of PML is confirmed.
The physician should be particularly alert to symptoms suggestive of PML that the patient may not notice (e.g. cognitive, neurological, or psychiatric symptoms).
Pulmonary toxicity. Cases of pulmonary toxicity including pneumonitis, interstitial lung disease, and acute respiratory distress syndrome (ARDS), some with fatal outcomes, have been reported in patients receiving Adcetris. Although a causal association with Adcetris has not been established, the risk of pulmonary toxicity cannot be ruled out.
Cases of pulmonary toxicity most commonly developed during the first 5 cycles of Adcetris and presented with cough, dyspnoea and interstitial lung infiltrates on radiological studies. Severity has been variable, with increased severity more likely with early onset. Some cases in which pneumonitis developed after the first cycle of Adcetris have had a fulminant course, requiring mechanical ventilation, treatment with high dose systemic corticosteroids and discontinuation of Adcetris. Some of these cases have had fatal outcome despite these measures; in others, the pneumonitis has resolved and treatment with Adcetris was resumed without recurrence of pneumonitis. Non-serious cases were more likely to occur after 3-5 cycles. These have been variably managed with Adcetris continued or dose delay or discontinuation. Corticosteroids were not administered and in many of these patients the pneumonitis did not resolve.
In the event of new or worsening symptoms that are rapidly progressive or occur in the first one to two cycles, Adcetris should be with-held and treatment with systemic corticosteroids commenced. If full resolution occurs, resumption of Adcetris treatment may be considered. If the presentation is non-serious with onset after several cycles of Adcetris, management may be by dose delay. Systemic corticosteroids may be considered.
Pancreatitis. Acute pancreatitis has been observed in patients treated with Adcetris. Fatal outcomes have been reported. Patients should be closely monitored for new or worsening abdominal pain, which may be suggestive of acute pancreatitis. Patient evaluation may include physical examination, laboratory evaluation for serum amylase and serum lipase, and abdominal imaging, such as ultrasound and other appropriate diagnostic measures. Adcetris should be held for any suspected case of acute pancreatitis.
Adcetris should be discontinued if a diagnosis of acute pancreatitis is confirmed.
Serious infections and opportunistic infections. Serious infections such as pneumonia, staphylococcal bacteraemia, sepsis/ septic shock (including fatal outcomes) and herpes zoster, cytomegalovirus (CMV) (reactivation) and opportunistic infections such as Pneumocystis jirovecii pneumonia and oral candidiasis have been reported in patients treated with Adcetris. Patients should be carefully monitored during treatment for the emergence of possible serious and opportunistic infections.
Infusion-related reactions. Immediate and delayed infusion-related reactions (IRR), as well as anaphylaxis, have been reported. Patients should be carefully monitored during and after infusion. If anaphylaxis occurs, administration of Adcetris should be immediately and permanently discontinued and appropriate medical therapy should be administered. If an infusion-related reaction occurs, the infusion should be interrupted and appropriate medical management instituted. The infusion may be restarted at a slower rate after symptom resolution. Patients who have experienced a prior infusion-related reaction should be pre-medicated for subsequent infusions. Premedication may include paracetamol, an antihistamine and a corticosteroid.
Infusion-related reactions are more frequent and more severe in patients with antibodies to Adcetris (see Section 4.8 Adverse Effects (Undesirable Effects)).
Tumour lysis syndrome. Tumour lysis syndrome (TLS) has been reported with Adcetris. Patients with rapidly proliferating tumour and high tumour burden are at risk of tumour lysis syndrome. These patients should be monitored closely and managed according to best medical practice. Management of TLS may include aggressive hydration, monitoring of renal function, correction of electrolyte abnormalities, anti-hyperuricaemic therapy, and supportive care.
Peripheral neuropathy. Adcetris treatment may cause a peripheral neuropathy, both sensory and motor. Adcetris-induced peripheral neuropathy is typically an effect of cumulative exposure to this medicinal product and is reversible in most cases with a 16-week median time from onset to resolution.
In clinical trials, the majority of patients had improvement or resolution of some of their symptoms (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for symptoms of neuropathy, such as hypoesthesia, hyperesthesia, paraesthesia, discomfort, a burning sensation, neuropathic pain or weakness. Patients experiencing new or worsening peripheral neuropathy may require a delay and a dose reduction of Adcetris or discontinuation of treatment (see Section 4.2 Dose and Method of Administration).
Haematological toxicities. Grade 3 or grade 4 anaemia, thrombocytopenia, and prolonged (≥ 1 week) grade 3 or grade 4 neutropenia can occur with Adcetris. Complete blood counts should be monitored prior to administration of each dose. If grade 3 or grade 4 neutropenia develops, manage as needed by dose modifications or discontinuations (see Section 4.2 Dose and Method of Administration).
Febrile neutropenia. Febrile neutropenia (fever of unknown origin without clinically or microbiologically documented infection with an absolute neutrophil count < 1.0 x 109/L, fever ≥ 38.5°C; ref Common Terminology Criteria for Adverse Events (CTCAE) v3) has been reported with treatment with Adcetris. Complete blood counts should be monitored prior to administration of each dose of this treatment. Patients should be monitored closely for fever and managed according to best medical practice if febrile neutropenia develops.
In combination therapy with AVD, CHP or as the BrECADD regimen, advanced age was a risk factor for febrile neutropenia. When Adcetris is administered in combination with AVD or CHP, primary prophylaxis with G-CSF, beginning with the first dose, is recommended for all patients regardless of age. When Adcetris is administered in combination as part of the BrECADD regimen, primary prophylaxis with G-CSF must be given beginning on day 5 of each cycle for all adult patients regardless of age.
Severe cutaneous adverse reactions (SCARs). Cases of SCARs, including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN) and drug reaction with eosinophilia and systemic symptoms (DRESS) have been reported with Adcetris. Fatal outcomes have been reported for SJS and TEN. If SJS, TEN or DRESS occur, treatment with Adcetris should be discontinued and appropriate medical therapy should be administered.
Gastrointestinal complications. Serious gastrointestinal (GI) complications including intestinal obstruction, ileus, enterocolitis, neutropenic colitis, erosion, ulcer, perforation and haemorrhage, some with fatal outcomes, have been reported in patients treated with Adcetris. Some cases of GI perforations were reported in lymphoma patients with pre-existing GI involvement. In the event of new or worsening GI symptoms, consider withholding Adcetris, perform a prompt diagnostic evaluation and treat appropriately.
Hepatotoxicity. Cases of hepatoxicity, ranging from asymptomatic elevations in serum transaminase levels to hepatic failure and fulminant hepatitis have been reported in patients receiving Adcetris. These have included fatal outcomes. Pre-existing liver disease, comorbidities, and concomitant medications may increase the risk of serious or fatal hepatotoxicity.
Hepatotoxicity most commonly presents as asymptomatic minor elevations in transaminases, although cholestasis has also been reported. In most patients with minor elevations, Adcetris was continued and the elevated transaminase levels resolved. Dose delay or reduction or discontinuation has also been described. The use of immunosuppressive drugs, including corticosteroids in the treatment of hepatitis associated with Adcetris has not been described.
Liver function should be tested before initiating the treatment and with every cycle during treatment with Adcetris. Depending on the severity of the hepatotoxicity event, consider delaying, reducing the dose or discontinuing Adcetris (see Section 4.2 Dose and Method of Administration).
Hyperglycaemia. Hyperglycaemia has been reported during clinical trials in patients with an elevated body mass index (BMI) with or without a history of diabetes mellitus. However, any patient who experiences an event of hyperglycaemia should have their serum glucose closely monitored. Anti-diabetic treatment should be administered as appropriate.
Long-term safety. There is insufficient evidence to judge the safety of treatment extended past 12 months.
Use in hepatic impairment. An excretion study found that approximately 75% of the clearance of unchanged MMAE is by excretion in the faeces. This is thought to be due to biliary excretion. The liver is a major route of elimination of the unchanged active metabolite MMAE. Limited clinical data from patients who were administered 1.2 mg/kg of Adcetris suggest that exposure to MMAE increased approximately 2.3-fold in patients with any degree of hepatic impairment. Adcetris should be used with caution in patients with hepatic impairment. Patients should be closely monitored for adverse events. Treatment with Adcetris should be discontinued in patients with hepatic impairment who are not demonstrating an adequate response to treatment.
Use in renal impairment. An excretion study found that approximately 25% of the clearance of unchanged MMAE is by excretion in the urine. A study evaluated the PK of brentuximab vedotin and MMAE after the administration of 1.2 mg/kg of Adcetris to patients with mild (n = 4), moderate (n = 3) and severe (n = 3) renal impairment. Patients requiring dialysis were excluded from this study. Compared to patients with normal renal function, MMAE exposure increased approximately 1.9-fold in patients with severe renal impairment (creatinine clearance < 30 mL/min). Adcetris should be used with caution in patients with renal impairment. Patients should be closely monitored for adverse events (see Section 4.2 Dose and Method of Administration).
Use in the elderly. Based upon population PK analyses and the similar safety profile in elderly patients with CTCL, no dosage adjustments are required in patients aged 65 and older (see Section 5.2 Pharmacokinetic Properties, Pharmacokinetics in special populations, Elderly patients).
The safety and efficacy of Adcetris as part of the BrECADD regimen has not been established in patients over the age of 60 years.
Paediatric use. The safety and efficacy of Adcetris in children less than 18 years have not yet been established. Clinical studies of Adcetris did not include sufficient numbers of subjects less than or younger than 18 years of age to determine whether they respond differently from adult subjects aged 18 years or older. Findings from nonclinical studies that may be relevant to the paediatric population were lymphoid depletion and reduced thymic weight. These observations in nonclinical studies were consistent with the pharmacologic disruption of microtubules caused by MMAE derived from Adcetris.
Effects on laboratory tests. No data available.
4.5 Interactions with Other Medicines and Other Forms of Interactions
The combined use of Adcetris and bleomycin is associated with pulmonary toxicity and is therefore contraindicated (see Section 4.3 Contraindications).
There are no drug-drug interactions data available with other chemotherapy regimens.
Interaction with medicinal products metabolised through CYP3A4 route (CYP3A4 inhibitors/ inducers). Coadministration of Adcetris with ketoconazole, a strong CYP3A4 and P-gp inhibitor, increased the exposure to the antimicrotubule agent MMAE by approximately 73%, and did not alter the plasma exposure to Adcetris. Therefore, coadministration of Adcetris with strong CYP3A4 and P-gp inhibitors may increase the incidence of neutropenia. If neutropenia develops, see Section 4.2 Dose and Method of Administration.
Coadministration of Adcetris with rifampicin, a strong CYP3A4 inducer, did not alter the plasma exposure to brentuximab vedotin; however, it reduced exposure to MMAE by approximately 31%.
Coadministration of midazolam, a CYP3A4 substrate, with Adcetris did not alter the metabolism of midazolam; therefore, Adcetris is not expected to alter the exposure to medicines that are metabolised by CYP3A4 enzymes.
Doxorubicin, vinblastine and dacarbazine. The serum and plasma pharmacokinetic characteristics of antibody drug conjugate and MMAE, respectively, following administration of brentuximab vedotin in combination with AVD were similar to that in monotherapy.
Co-administration of Adcetris did not affect the plasma exposure of AVD.
Cyclophosphamide, doxorubicin, and prednisone. The serum and plasma pharmacokinetic characteristics of antibody drug conjugate and MMAE, respectively, following administration of brentuximab vedotin in combination with CHP were similar to that in monotherapy.
Etoposide, cyclophosphamide, doxorubicin, dacarbazine, dexamethasone (BrECADD). The pharmacokinetics of ADC and MMAE have not been characterised in the setting of BrECADD. Exposures of brentuximab vedotin and concurrent chemotherapy are not expected to be affected in the BrECADD regimen.
Coadministration with other CYP substrates. In vitro studies indicated that clinical drug-drug interactions are unlikely to occur as a result of MMAE-mediated inhibition or induction of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 or CYP2D6.
Coadministration with drugs that are substrates of transporters. In vitro, MMAE was not a substrate for the BCRP, MRP2, OATP1B1, OATP1B3, OAT1 or OCT2 transporters.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. The effects of Adcetris on human male and female fertility have not been studied.
However, results from toxicity studies in rats indicate the potential for brentuximab vedotin to impair male reproductive function and fertility. Seminiferous tubule degeneration, sertoli cell vacuolation, reduced spermatogenesis and aspermia were observed in male rats that received weekly IV injections of ≥ 5 mg/kg brentuximab vedotin. The no-effect dose (0.5 mg/kg) is below the recommended human dose of 1.8 mg/kg based on bodyweight. Testicular atrophy and degeneration had not fully reversed following a 16 week treatment free period. MMAE, the main active metabolite of brentuximab vedotin, has been shown to have aneugenic properties in an in vivo rat bone marrow micronucleus study. These results were consistent with the pharmacological effect of MMAE on the mitotic apparatus (disruption of the microtubule network) in cells.
Therefore, men being treated with this medicine are advised to have sperm samples frozen and stored before treatment. Men being treated with this medicine are advised not to father a child during treatment and for up to 6 months following the last dose. Effects on spermatogenesis cannot be excluded after a 6-month treatment free period.
While not observed with brentuximab vedotin, ovarian effects were observed in repeat dose toxicity studies of other MMAE-containing ADCs. A mild to moderate decrease in, or absence of, secondary and tertiary ovarian follicles was observed in young female cynomolgus monkeys at doses ≥ 3 mg/kg weekly for 4 weeks. These effects showed evidence of recovery 6 weeks after the end of dosing and no changes were observed in primordial follicles.
Use in pregnancy. (Category D)
There are no adequate and well controlled studies with Adcetris in pregnant women. However, based on its mechanism of action and findings in animals, Adcetris can cause fetal harm when administered to a pregnant woman.
Embryofetal toxicities were seen in a rat embryofetal development study in which pregnant rats received two IV doses of ≥ 3 mg/kg brentuximab vedotin during a period of organogenesis, and included an increased incidence of postimplantation loss, decreased number of foetuses and an increase in the incidence of external malformations (umbilical hernias and malrotated hindlimbs).
Adcetris should not be used during pregnancy unless the benefit to the mother outweighs the potential risks to the fetus. If a pregnant woman needs to be treated, she should be clearly advised on the potential risk to the fetus. Women of childbearing potential should be using two methods of effective contraception during treatment with Adcetris and until 6 months after treatment.
See Effects on fertility section above pertaining to advice for women whose male partners are being treated with Adcetris.
Use in lactation. There are no data as to whether Adcetris or its metabolites are excreted in human milk. A risk to the newborn/infant cannot be excluded. A decision should be made whether to discontinue breastfeeding or to discontinue/abstain from this therapy, taking into account a potential risk of breastfeeding for the child and the benefit of therapy for the woman.
4.7 Effects on Ability to Drive and Use Machines
Brentuximab vedotin may have a minor influence on the ability to drive and use machines.
4.8 Adverse Effects (Undesirable Effects)
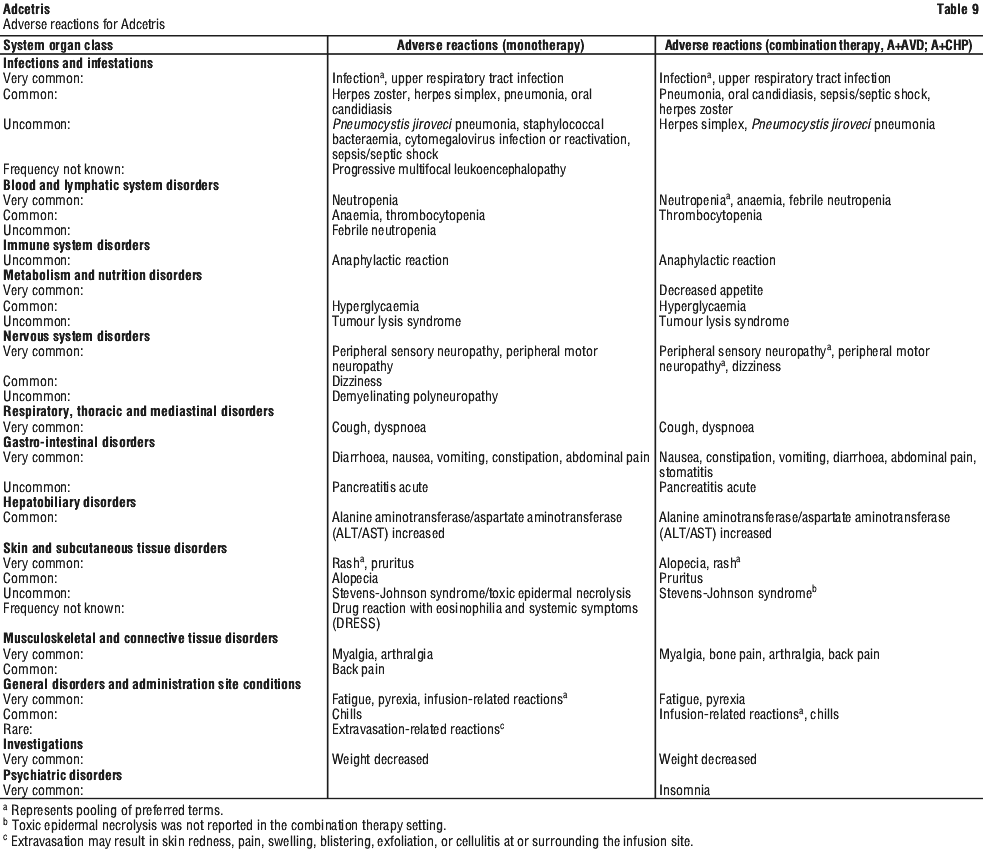
Summary of the safety profile. The safety profile of Adcetris is based on available clinical trial data, the named patient program (NPP), and post-marketing experience to date. Frequencies of adverse reactions described below and in Table 9 have been determined based on data generated from clinical studies.
Monotherapy. In the pooled dataset of Adcetris as monotherapy across HL, sALCL and CTCL studies (SG035 0003, SG035 0004, SG035 005, SG035 006, C25001, C25007) the most frequent adverse reactions (≥ 10%) were infections, peripheral sensory neuropathy, nausea, fatigue, diarrhoea, pyrexia, neutropenia, rash, cough and upper respiratory tract infection, vomiting, arthralgia, peripheral motor neuropathy, infusion-related reactions, pruritus, constipation, dyspnoea, weight decreased, myalgia and abdominal pain.
Serious adverse drug reactions occurred in 12% of patients. The frequency of unique serious adverse drug reactions was ≤ 1%. Adverse events led to the discontinuation of study treatment in 24% of patients receiving Adcetris.
The safety data in patients retreated with Adcetris (SGN35-006) were consistent with those observed in the combined pivotal phase 2 studies, with the exception of peripheral motor neuropathy, which had a higher incidence (28% vs. 9% in the pivotal phase 2 studies) and was primarily grade 2. Patients also had a higher incidence of arthralgia, grade 3 anaemia, and back pain compared to patients observed in the combined pivotal phase 2 studies.
The safety data reported from the phase 1 dose escalation and clinical pharmacology studies (n = 15 patients) and from the Named Patient Program (NPP; n = 26 patients), in patients with relapsed or refractory HL who had not received an ASCT (see Section 5.1 Pharmacodynamic Properties, Clinical trials), and were treated with the recommended dose of 1.8 mg/kg every three weeks, were consistent with the safety profile of the pivotal clinical studies.
Combination therapy. For safety information of agents given in combination with Adcetris (doxorubicin, vinblastine and dacarbazine [AVD]; or cyclophosphamide, doxorubicin, and prednisone [CHP]; or etoposide, cyclophosphamide, doxorubicin, dacarbazine, dexamethasone [BrECADD]), refer to their product information.
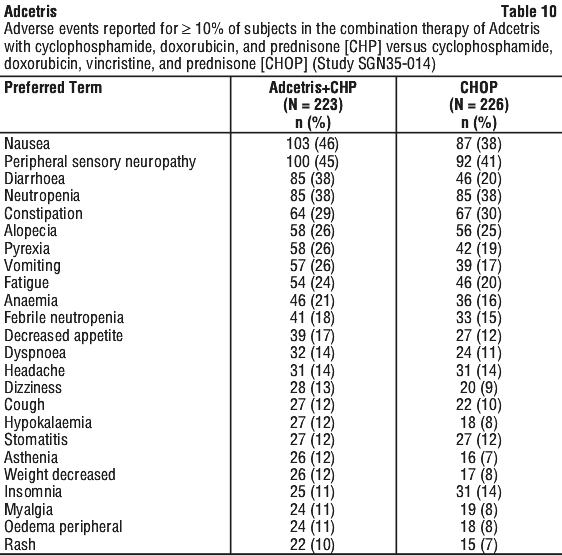
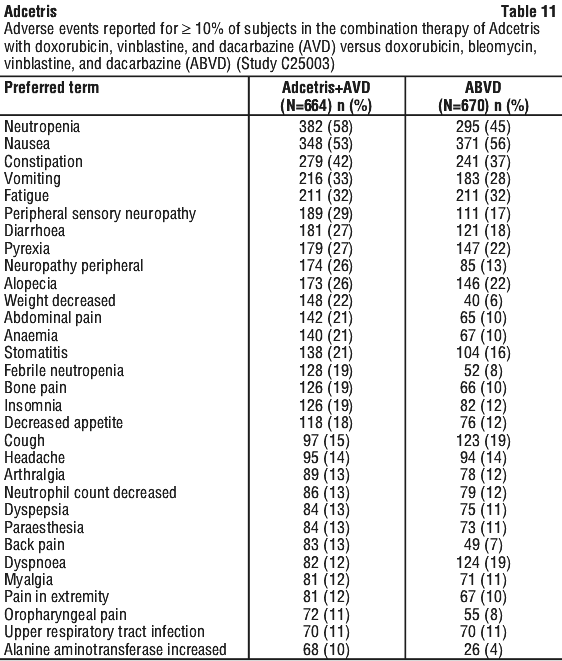
Combination therapy (Adcetris + AVD; Adcetris + CHP). In the studies of Adcetris as combination therapy with AVD in 662 patients with previously untreated HL (C25003) and with CHP in 223 patients with previously untreated PTCL (SGN35-014), the most common adverse reactions (≥ 10%) were: infections, neutropenia, peripheral sensory neuropathy, nausea, constipation, vomiting, diarrhoea, fatigue, pyrexia, alopecia, anaemia, weight decreased, stomatitis, febrile neutropenia, abdominal pain, decreased appetite, insomnia, bone pain, rash, cough, dyspnoea, arthralgia, myalgia, back pain, peripheral motor neuropathy, upper respiratory tract infection, and dizziness.
In patients receiving Adcetris combination therapy, serious adverse reactions occurred in 34% of patients. Serious adverse reactions occurring in ≥ 3% of patients included febrile neutropenia (15%), pyrexia (5%), infection (11%), and neutropenia (3%).
Adverse events led to treatment discontinuation in 10% of patients. Adverse events that led to treatment discontinuation in ≥ 2% of patients included peripheral sensory neuropathy and peripheral neuropathy.
Tabulated list of adverse reactions. Adverse reactions for Adcetris are listed by MedDRA system organ class and preferred term (see Table 9). Within each system organ class, adverse reactions are listed under frequency categories of: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000); frequency not known (cannot be estimated from the available data).
Combination therapy (BrECADD). In the HD21 study, 747 patients received BrECADD, and 741 patients received eBEACOPP (escalated bleomycin [B], etoposide [E], doxorubicin [A], cyclophosphamide [C], vincristine [O], procarbazine [P] and prednisone [P]).
Serious adverse reactions occurred in 39.4% patients receiving BrECADD treatment, and 36.4% in patients who received eBEACOPP. The most common serious adverse reactions in patients who received BrECADD (> 3%) were febrile neutropenia (19.3%), pyrexia (3.9%), neutropenia (3.2%).
Serious adverse events led to treatment discontinuation in 2% of patients in both BrECADD and eBEACOPP arms. The most common serious adverse event that led to discontinuation in the BrECADD arm were febrile neutropenia (0.3%) and cardiac failure (0.3%).
The most common treatment-emergent adverse events (≥ 10%) for BrECADD versus eBEACOPP are listed in Table 12.
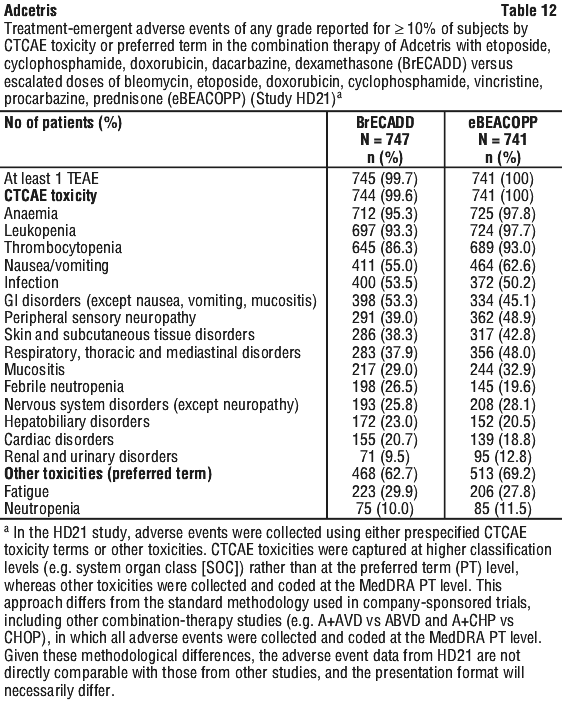
Severe and prolonged (≥ 1 week) neutropenia can occur with this treatment which may increase the risk of patients developing serious infections. Febrile neutropenia was reported in < 1% of the patients.
In the pivotal phase 2 population (SG035-0003 and SG035-0004), the median duration of grade 3 or grade 4 neutropenia was limited (1 week); 2% of patients had grade 4 neutropenia that lasted ≥ 7 days. Less than half of the patients in the phase 2 population with grade 3 or grade 4 neutropenia had temporally associated infections, and the majority of temporally associated infections were grade 1 or grade 2.
Combination therapy. In the clinical trials C25003 (Adcetris + AVD) and SGN35-014 (Adcetris + CHP) of Adcetris as combination therapy, neutropenia led to dose delays in 19% of patients. Grade 3 neutropenia was reported in 17% and grade 4 neutropenia was reported in 41% of patients. Two percent of patients required dose reduction and < 1% of patients discontinued one or more of the study drugs due to neutropenia.
Febrile neutropenia was reported in 20% of the patients who did not receive primary prophylaxis with G-CSF (see Section 4.2). The frequency of febrile neutropenia was 13% in patients who received primary prophylaxis with G-CSF.
In the HD21 clinical trial (BrECADD) of Adcetris as combination therapy, neutropenia led to dose delays in 0.5% of patients. Grade 3 neutropenia was reported in 0.5% and grade 4 neutropenia was reported in 9% of patients. A dose reduction was required in 1.1% of patients and there was no treatment discontinuation due to serious neutropenia. All patients received primary prophylaxis with G-CSF (see Section 4.2 Dose and Method of Administration). The frequency of febrile neutropenia was 26.5% in patients who received BrECADD.
Serious infections and opportunistic infections. Monotherapy. In clinical trials, serious infections and opportunistic infections occurred in 10% of patients, sepsis or septic shock occurred in < 1% of the patients. The most commonly reported opportunistic infections were herpes zoster and herpes simplex.
PML has been reported outside of the pivotal clinical trials described in this section (see Section 4.4 Special Warnings and Precautions for Use).
Combination therapy. In the clinical trials (C25003 [Adcetris + AVD] and SGN35-014 [Adcetris + CHP]) of Adcetris as combination therapy, serious infections, including opportunistic infections occurred in 15% of patients; sepsis, septic shock or bacteraemia occurred in 4% of the patients. The most commonly reported opportunistic infections were herpes viral infections.
In the clinical trial (HD21 [BrECADD]) of Adcetris as combination therapy, serious infections and infestations occurred in 14.3% of patients. The most commonly reported events were infection (2%), pneumonia (1.7%), and neutropenic infection (1.2%).
Peripheral neuropathy. Monotherapy. In clinical trials treatment emergent neuropathy occurred in 59% of the population, peripheral motor neuropathy occurred in 14% of patients. Peripheral neuropathy led to treatment discontinuation in 15%, dose reductions in 15%, and dose delays in 17% of patients. For patients who experienced peripheral neuropathy the median time of onset of peripheral neuropathy was 12 weeks. The median duration of treatment for patients who discontinued due to peripheral neuropathy was 12 cycles.
Among patients who experienced peripheral neuropathy in the pivotal phase 2 studies (SG035-0003 and SG035-0004) and randomised phase 3 studies (SGN35-005 and C25001), the median follow-up time from end of treatment until last evaluation ranged from 48.9 to 98 weeks. At the time of last evaluation, 82-85% of patients who experienced peripheral neuropathy had resolution or improvement of their peripheral neuropathy symptoms. The median time from onset to resolution or improvement for all events ranged from 16 to 23.4 weeks.
In patients with relapsed or refractory HL or relapsed or sALCL who were retreated with Adcetris (SGN35-006), the majority of patients (80%) also had improvement or resolution of their peripheral neuropathy symptoms at the time of last evaluation.
Combination therapy. In the clinical trial of Adcetris as combination therapy with AVD, treatment emergent neuropathy occurred in 67% of the population; peripheral motor neuropathy occurred in 11% of patients. Peripheral neuropathy led to treatment discontinuation in 7%, dose reductions in 21%, and dose delays in 1% of patients. For patients who experienced peripheral neuropathy the median time of onset of peripheral neuropathy was 8 weeks. Patients who discontinued due to peripheral neuropathy received a median of 8 doses of Adcetris + AVD (A+AVD) before discontinuation of one or more agents.
Among patients who experienced peripheral neuropathy, the median follow up time from end of treatment until last evaluation was approximately 286 weeks. At the time of last evaluation, most of the patients (86%) who experienced peripheral neuropathy had resolution or improvement of their peripheral neuropathy symptoms. The median time from onset to resolution or improvement of peripheral neuropathy events was 17 weeks (ranged from 0 weeks to 283 weeks).
In the clinical trial of Adcetris as combination therapy with CHP, treatment emergent neuropathy occurred in 52% of the population; peripheral motor neuropathy occurred in 9% of patients. Peripheral neuropathy led to treatment discontinuation in 1%, dose reductions in 7% and dose delays in < 1% of patients. For patients who experienced peripheral neuropathy the median time of onset was 9.1 weeks. Patients who discontinued due to peripheral neuropathy received a median of 5 doses of Adcetris + CHP (A+CHP) before discontinuation of one or more agents.
Among patients who experienced peripheral neuropathy, the median follow up time from end of treatment until last evaluation was approximately 177 weeks. At the time of last evaluation, 64% who experienced peripheral neuropathy had resolution or improvement of their peripheral neuropathy symptoms. The median time from onset to resolution or improvement of peripheral neuropathy events was 19 weeks (ranged from 0 weeks to 195 weeks).
In the clinical trial (HD21 [BrECADD]) of Adcetris as combination therapy, treatment emergent peripheral sensory neuropathy occurred in 38.8% of the population; peripheral motor neuropathy occurred in 3.6% of patients. Peripheral sensory neuropathy as a serious adverse event did not lead to discontinuation in any patients, led to dose reduction in 4.7% and treatment delay in 1.5% of patients. Peripheral motor neuropathy as a serious adverse event did not lead to discontinuation in any patients, led to dose reduction in 0.7%, and treatment delay in 0.1% of patients.
Acute pancreatitis. Acute pancreatitis (including fatal outcomes) has been reported outside of the pivotal clinical trials. Consider the diagnosis of acute pancreatitis for patients presenting with new or worsening abdominal pain (see Section 4.4 Special Warnings and Precautions for Use).
Infusion-related reactions. Monotherapy. Infusion-related reactions such as headache, rash, back pain, vomiting, chills, nausea, dyspnoea, pruritus, and cough were reported in 13% of patients.
Anaphylaxis has been reported. Symptoms of anaphylaxis may include, but are not limited to, urticaria, angioedema, hypotension and bronchospasm.
Combination therapy. In clinical trials (C25003 [Adcetris + AVD] and SGN35-014 [Adcetris + CHP]), infusion-related reactions, such as headache, rash, back pain, vomiting, chills, nausea, dyspnoea, pruritus, cough, infusion site pain and pyrexia were reported in 8% of patients. Dizziness, extravasation, hypoaesthesia, and hypoxia were reported in 4% of patients. Symptoms of an anaphylactic reaction may include, but are not limited to, urticaria, angioedema, hypotension and bronchospasm.
Immunogenicity. In clinical trials, patients were periodically tested for antibodies to brentuximab vedotin using a sensitive electrochemiluminescent immunoassay. There was a higher incidence of infusion-related reactions observed in patients with persistently positive antibodies to brentuximab vedotin relative to patients who tested transiently positive or negative.
The presence of antibodies to brentuximab vedotin did not correlate with a clinically meaningful reduction in serum brentuximab vedotin levels and did not result in a decrease in the efficacy of Adcetris.
Immunogenicity assay results are highly dependent on several factors including assay sensitivity and specificity, assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to brentuximab vedotin with the incidence of antibodies to other products may be misleading.
Elderly. Combination therapy. In older patients from clinical trials C25003 [Adcetris + AVD] and SGN35-014 [Adcetris + CHP] (≥ 60 years of age), the incidence of adverse events was similar across treatment arms. More serious adverse events and dose modifications (including dose delays, reductions, and discontinuations) were reported in the older patients compared with the overall study population. Advanced age was a risk factor for febrile neutropenia in patients in both arms. Older patients who received G-CSF primary prophylaxis had lower incidence of neutropenia and febrile neutropenia than those who did not receive G-CSF primary prophylaxis.
The safety and efficacy of Adcetris as part of the BrECADD regimen have not been established in patients over the age of 60 years.
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
4.9 Overdose
There is no known antidote for overdose of Adcetris. In case of overdose, the patient should be closely monitored for adverse reactions, particularly neutropenia, and supportive treatment should be administered (see Section 4.4 Special Warnings and Precautions for Use).
For information on the management of overdose, contact the Poison Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action. Brentuximab vedotin is an ADC that delivers an antineoplastic agent that results in apoptotic cell death in CD30-expressing tumour cells (such as classical Hodgkin's lymphoma and systemic anaplastic large cell lymphoma). Nonclinical data suggest that the biological activity of brentuximab vedotin results from a multi-step process. Binding of the ADC to CD30 on the cell surface initiates internalisation of the ADC-CD30 complex, which then traffics to the lysosomal compartment. Within the cell, a single defined active species, MMAE, is released via proteolytic cleavage. Binding of MMAE to tubulin disrupts the microtubule network within the cell, induces cell cycle arrest and results in apoptotic death of the CD30-expressing tumour cell.
Contributions to the mechanism of action by other antibody associated functions have not been excluded.
Cardiac electrophysiology. Forty-six (46) patients with CD30 expressing hematologic malignancies were evaluable of the 52 patients who received 1.8 mg/kg of brentuximab vedotin every 3 weeks as part of a phase 1, single arm, open label, multicentre cardiac safety study. The primary objective was to evaluate the effect of brentuximab vedotin on cardiac ventricular re-polarization and the predefined primary analysis was the change in QTc from baseline to multiple time points in cycle 1.
The upper 90% confidence interval (CI) around the mean effect on QTc was < 10 msec at each of the cycle 1 and cycle 3 post-baseline time points. These data indicate the absence of clinically relevant QT prolongation due to brentuximab vedotin administered at a dose of 1.8 mg/kg every 3 weeks in patients with CD30 expressing malignancies.
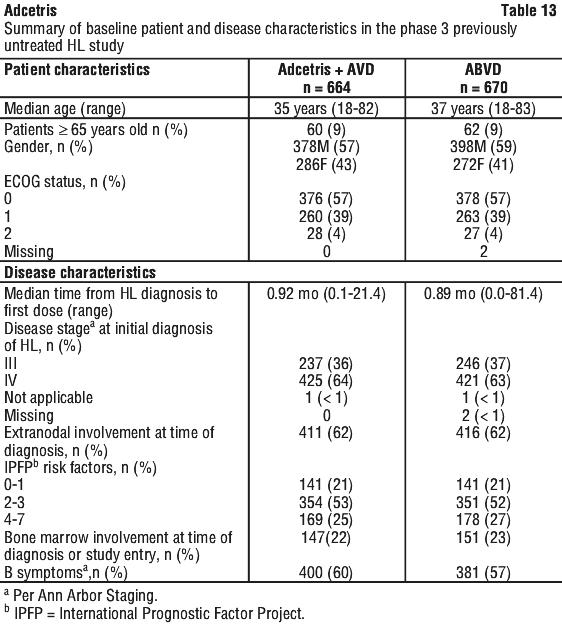
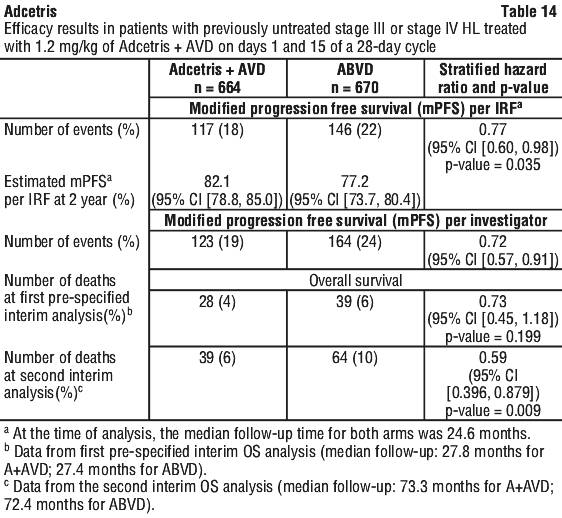
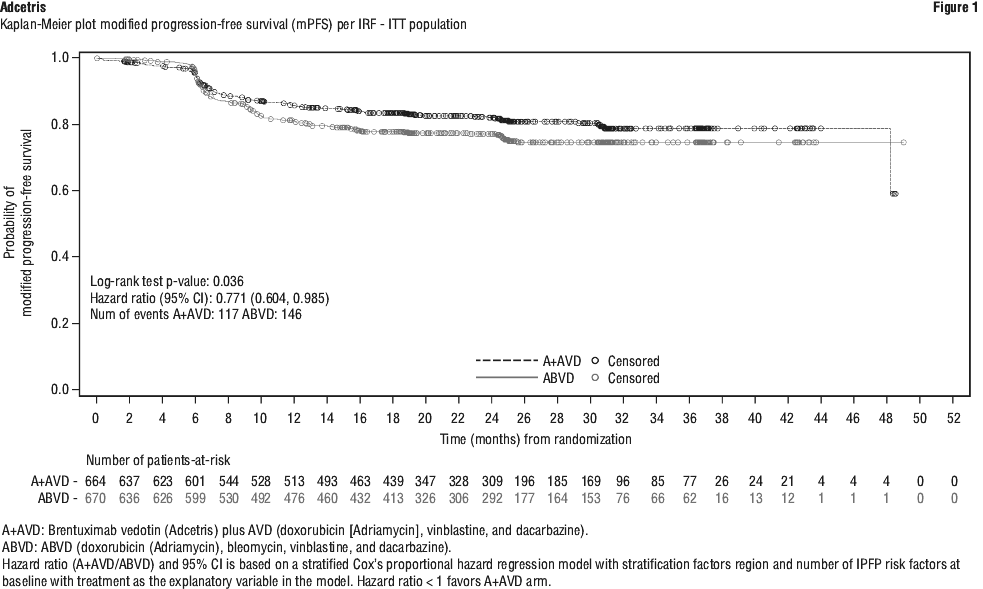
Clinical trials. Hodgkin lymphoma (HL). Patients with previously untreated CD30+ stage III or stage IV Hodgkin lymphoma (HL) in combination with doxorubicin [A], vinblastine [V], and dacarbazine [D] (AVD) (study C25003). The efficacy and safety of Adcetris were evaluated in a randomised, open-label, 2-arm, multicentre trial in 1334 patients with previously untreated stage III or stage IV HL in combination with chemotherapy AVD. All patients had CD30-expressing HL. Sixty-two percent of patients had extranodal site involvement. Of the 1334 patients, 664 patients were randomised to the Adcetris + AVD arm and 670 patients were randomised to the ABVD (doxorubicin [A], bleomycin [B], vinblastine [V] and dacarbazine [D]) arm and stratified by the number of International Prognostic Factor Project (IPFP) risk factors and region. Patients were treated with 1.2 mg/kg of Adcetris administered as an intravenous infusion over 30 minutes on days 1 and 15 of each 28-day cycle + AVD. The median number of cycles received was 6 (range, 1 to 6 cycles). Table 13 provides a summary of the baseline patient and disease characteristics.
Approximately one-third fewer patients treated with Adcetris + AVD received subsequent salvage chemotherapy (n = 66) and high-dose chemotherapy and transplant (n = 36) compared with those treated with ABVD (n = 99 and n = 54, respectively).
Subgroup analyses. Pre-specified subgroup analyses of modified PFS per IRF were performed for the ITT population including age, region, cancer stage at baseline, baseline extranodal sites, number of IPFP risk factors, baseline B symptoms, cycle 2 PET assessment, cycle 2 PET Deauville score, and receipt of alternative first line medication (AFM). The analyses showed a consistent trend towards benefit for patients who received Adcetris + AVD compared with patients who received ABVD in most subgroups. The efficacy in elderly patient population (patients ≥ 60 years of age [n = 186] [HR = 1.00, 95% CI (0.58, 1.72)] and ≥ 65 years of age [n = 122] [HR = 1.01, 95% CI (0.53, 1.94)]) and patients with no extranodal sites (n = 445) (HR = 1.04, 95% CI [0.67, 1.62]) showed no clinically meaningful difference between the two arms.
Overall survival results in the stage III population for patients treated with Adcetris + AVD compared with patients treated with ABVD showed a HR = 0.86, 95% CI, (0.452; 1.648). Overall survival results in the stage IV population indicated a 52% reduction in the risk of death for patients treated with Adcetris + AVD compared with patients treated with ABVD [HR = 0.48, 95% CI (0.286, 0.799)].
Adult patients with previously untreated CD30+ stage IIB with large mediastinal mass and/or extranodal disease, stage III or stage IV HL in combination with etoposide [E], cyclophosphamide [C], doxorubicin [A], dacarbazine [D], dexamethasone [D] (BrECADD) (Study HD21). The safety and efficacy of Adcetris were evaluated in an open-label, prospective, multicentre phase III trial in 1500 patients with previously untreated stage IIB with large mediastinal mass (mediastinal thoracic ratio [MTR] > 0.33) and/or extranodal lesions, stage III, or stage IV HL in combination with chemotherapy (etoposide [E], cyclophosphamide [C], doxorubicin [A], dacarbazine [D], dexamethasone [D] [BrECADD]). Of the 1500 patients, 751 patients were randomised to the BrECADD arm and 749 patients were randomised to eBEACOPP (escalated bleomycin [B], etoposide [E], doxorubicin [A], cyclophosphamide [C], vincristine [O], procarbazine [P] and prednisone [P]) arm and stratified by region of enrolment, age, sex, and international prognostic score (IPS). Patients in the BrECADD arm were treated on day 1 of each 21 day cycle with 1.8 mg/kg of Adcetris administered as an intravenous infusion over 30 minutes. Patients also received chemotherapy including cyclophosphamide 1250 mg/m2, doxorubicin 40 mg/m2, etoposide or etoposide phosphate 150 mg/m2, dacarbazine 250 mg/m2, and dexamethasone 40 mg.
All treated patients received primary prophylaxis with G-CSF (see Section 4.2 Dose and Method of Administration). After 2 cycles of treatment, restaging by PET was performed with PET negative patients to receive a total of 4 cycles and PET positive patients to receive a total of 6 cycles of therapy. The median number of cycles received in both arms was 4 (range, 1 to 6 cycles).
Table 15 provides a summary of the baseline patient and disease characteristics. There were no relevant differences in the patient and disease characteristics between the two arms.
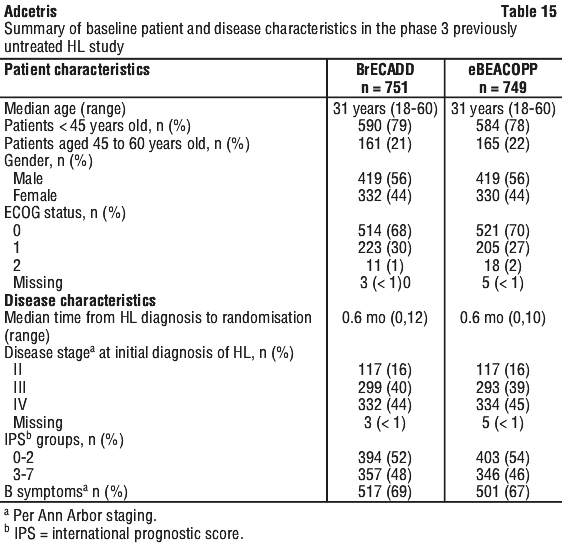
TRMB was defined as any grade 3 or grade 4 Common Terminology Criteria for Adverse Events (CTCAE) organ toxicity or grade 4 haematological toxicity during primary chemotherapy, including the period of up to 30 days after the last chemotherapy dose.
At the time of the primary analysis, superiority was met for TRMB with BrECADD with an absolute risk reduction of -16.7 percentage points and with a statistically significant reduction in relative risk (stratified RR (95% CI): 0.712 (0.643, 0.789). The coprimary endpoint PFS regardless of missed visits and regardless of initialisation of new anticancer therapy met noninferiority with a statistically significant reduction in risk in the BrECADD treatment arm compared with eBEACOPP (unstratified HR=0.62 [multiplicity adjusted 95% CI, 0.369, 1.040]) (data cut off 31 December 2022).
Table 16 provides TRMB by treatment arm.
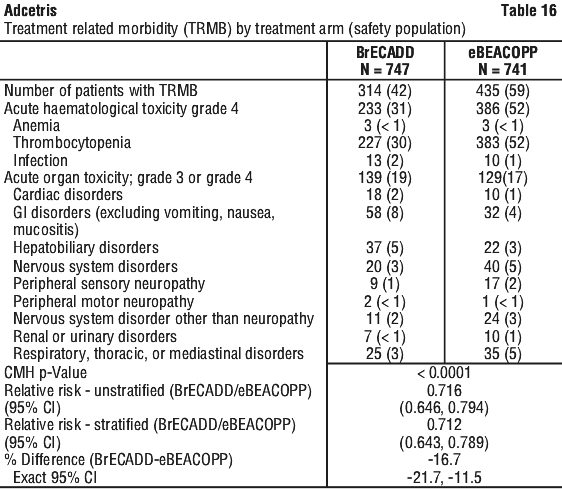
Table 17 provides the efficacy results for PFS in the ITT population as of a data cut off of 31 December 2022.
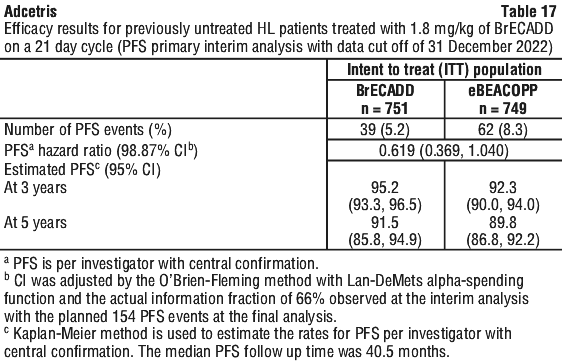
Prespecified subgroup analyses of PFS per investigator assessment with central confirmation were performed for the ITT population including age, sex, enrolment region, IPS score, baseline Ann Arbor disease stage, baseline ECOG, baseline B symptoms, and cycle 2 PET assessment. The analyses showed consistent evidence of non-inferiority for BrECADD arm patients compared with eBEACOPP group patients.
Patients with relapsed or refractory CD30+ HL following autologous stem cell transplant (ASCT) (study SG035-0003). The efficacy and safety of Adcetris as a single agent was evaluated in a pivotal open-label, single-arm, multicentre study (study SG035-0003) in 102 patients with relapsed or refractory HL. Adcetris was administered at a starting dose of 1.8 mg/kg (IV) infusion on day 1 of each 21-day cycle. See Table 18 for a summary of baseline patient and disease characteristics.
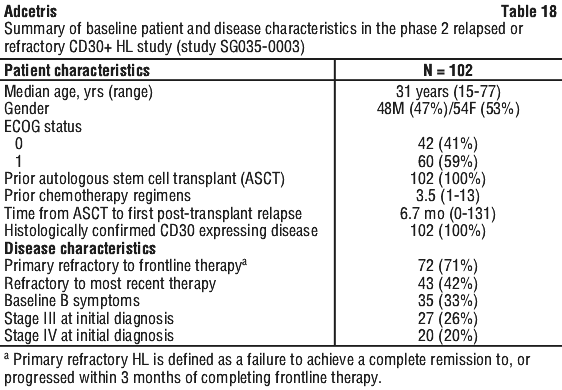
The objective response rate (ORR) per IRF assessment was 75% (76 of 102 patients in the intent to treat [ITT] set). Complete remission (CR) was 33% (34 of 102 patients in the ITT set). The median overall survival (OS) is 40.5 months (the median observation time (time to death or last contact) from first dose was 35.1 months). The estimated overall survival rate at 5 years was 41% (95% CI [31%, 51%]). The investigator assessments were generally consistent with the independent review of the scans. Of the patients treated, 8 responding patients went on to receive an allogeneic SCT. For further efficacy results see Table 19.
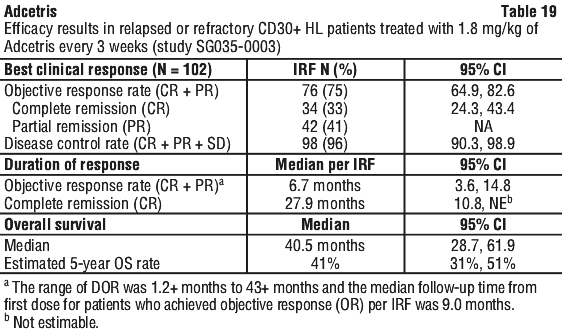
Tumour reduction was achieved in 94% of patients.
Of the 35 patients (33%) who had B symptoms at baseline, 27 patients (77%) experienced resolution of all B symptoms at a median time of 0.7 months from initiation of Adcetris.
Patients with relapsed or refractory CD30+ HL following at least two prior therapies when ASCT or multi-agent chemotherapy is not a treatment option. Data were collected from patients (n = 15) in phase 1 dose escalation and clinical pharmacology studies, and from patients (n = 26) in a Named Patient Program (NPP), with relapsed or refractory HL who had not received an ASCT, and who were treated with 1.8 mg/kg of Adcetris every 3 weeks.
Baseline patient characteristics showed failure from multiple prior chemotherapy regimens (median of 3 with a range of 1 to 7) before first administration with brentuximab vedotin. Fifty nine percent (59%) of patients had advanced stage disease (stage III or IV) at initial diagnosis.
Results from these phase 1 studies and from the NPP experience showed, that in patients with relapsed or refractory HL without prior ASCT, clinically meaningful responses can be achieved as evidenced by an investigator assessed, objective response rate of 54% and a complete remission rate of 22% after a median of 5 cycles of Adcetris.
Study SGN35-006 (retreatment study). The efficacy of retreatment in patients who had previously responded (CR or PR) to treatment with Adcetris was evaluated in a phase 2, open-label, multicentre trial. Twenty patients with relapsed or refractory HL received a starting dose of 1.8 mg/kg and one patient received a starting dose of 1.2 mg/kg of Adcetris administered intravenously over 30 minutes every 3 weeks. The median number of cycles was 7 (range, 2 to 37 cycles). Of the 20 evaluable patients with HL, 6 patients (30%) achieved a CR and 6 patients (30%) achieved a PR with Adcetris retreatment, for an ORR of 60%. The median duration of response was 9.2 and 9.4 months in patients who achieved OR (CR+PR) and CR, respectively.
Patients with CD30+ HL at risk of relapse or progression following ASCT (study SGN35-005). The efficacy and safety of Adcetris were evaluated in a randomised, double blinded, placebo controlled, 2-arm multicentre trial in 329 patients with HL at risk of relapse or progression following ASCT. The regulatory approval of this indication was based on an improvement in progression free survival only; no improvement in overall survival has been demonstrated.
See Table 20 for a summary of baseline patient characteristics. Of the 329 patients, 165 patients were randomised to the treatment arm and 164 patients were randomised to the placebo arm. The safety population in the Adcetris arm (N = 167) included two additional patients who received at least one dose of Adcetris but were not randomised to the treatment arm. In the study, patients were to receive their first dose after recovery from ASCT (between days 30-45 following ASCT). Patients were treated with 1.8 mg/kg of Adcetris or matching placebo intravenously over 30 minutes every 3 weeks for up to 16 cycles. The median number of cycles received in both arms was 15 cycles.
In addition to other inclusion criteria, patients were also required to present with at least one of the following:
HL that was refractory to frontline treatment.
Relapsed or progressive HL that occurred < 12 months from the end of frontline treatment.
Extranodal involvement at time of pre-ASCT relapse, including extranodal extension of nodal masses into adjacent vital organs.
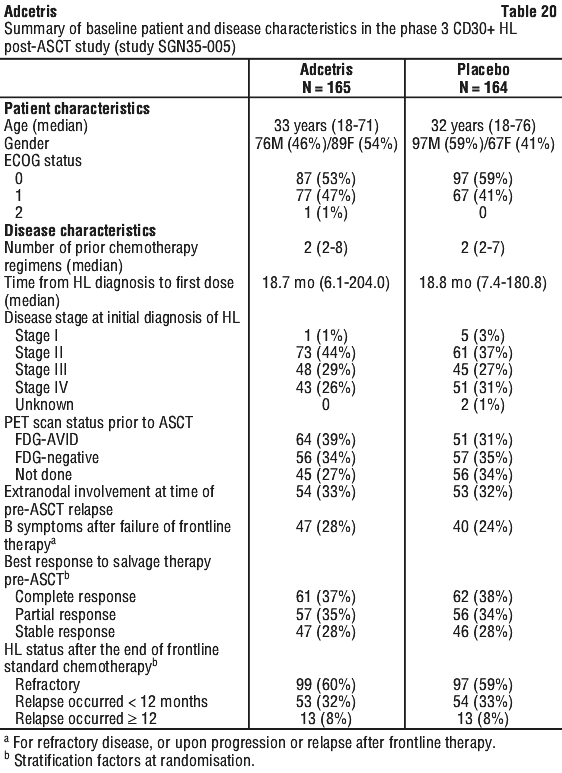

Quality of life was assessed using the EQ-5D instrument. No clinically meaningful differences were observed between the treatment and placebo arms. Eighty-five patients in the placebo arm progressed and received subsequent treatments, of whom 72 (84.7%) received Adcetris.
Peripheral T-cell lymphoma (PTCL). Patients with previously untreated CD30+ PTCL in combination with cyclophosphamide [C], doxorubicin [H], and prednisone [P] (CHP) (study SGN35-014). The efficacy and safety of Adcetris were evaluated in a randomised, double-blind, double-dummy, active-controlled, multicentre trial of 452 patients with previously untreated CD30+ PTCL in combination with cyclophosphamide [C], doxorubicin [H], and prednisone [P] (CHP). Of the 452 patients, 226 were randomised to treatment with Adcetris + CHP and 226 patients were randomised to treatment with CHOP (cyclophosphamide [C], doxorubicin [H], vincristine [O], and prednisone [P]). Randomisation was stratified by ALK-positive sALCL versus all other subtypes and by the International Prognostic Index (IPI) score. Patients were treated with 1.8 mg/kg of Adcetris administered as an intravenous infusion over 30 minutes on day 1 of each 21-day cycle for 6 to 8 cycles + CHP. The median number of cycles received was 6 (range, 1 to 8 cycles); 70% of patients received 6 cycles of treatment, and 18% received 8 cycles of treatment. Table 22 provides a summary of baseline patient and disease characteristics.
Key secondary endpoints included PFS per IRF for subjects with centrally-confirmed sALCL, CR rate per IRF following the completion of study treatment, OS, and ORR per IRF following the completion of study treatment which were tested by a fixed sequence testing procedure following the statistical significance of PFS per IRF.
The primary endpoint and alpha-protected, key secondary endpoints, which were evaluated hierarchically, were met. The median PFS per IRF was 48.2 months on the Adcetris + CHP arm versus 20.8 months on the CHOP arm. The stratified hazard ratio was 0.71 (95% CI: 0.54, 0.93, p = 0.011), indicating a 29% reduction in the risk of PFS events for Adcetris + CHP versus CHOP. Table 23 provides the efficacy results for PFS and other key secondary endpoints.
PFS per IRF for patients with centrally-confirmed sALCL was a pre-specified key secondary endpoint. The median PFS per IRF was 55.7 months on the Adcetris + CHP arm versus 54.2 months on the CHOP arm. The stratified hazard ratio was 0.59 (95% CI, 0.42; 0.84), compatible with a statistically significant 41% reduction in the risk of PFS events for Adcetris + CHP versus CHOP (p value = 0.003). No formal testing was performed in other histological subtypes, and no histological subtypes of PTCL were powered to detect a significant difference in treatment effect.

As of study closure more than 7 years after enrolment of the first patient, PFS per investigator results in the ITT population indicated a 30% reduction in the risk of a PFS event in the Adcetris + CHP arm compared with patients treated with CHOP (HR = 0.70 [95% CI (0.53, 0.91)]).
As of study closure, overall survival results continued to show a benefit and were consistent with those reported at the time of the primary analysis. Overall survival results in the ITT population indicated a 28% reduction in the risk of death in the Adcetris + CHP arm compared with patients treated with CHOP (HR = 0.72 [95% CI (0.53 to 0.99)]).
Patients with relapsed or refractory sALCL (study SG035-0004). The efficacy and safety of Adcetris as a single agent was evaluated in an open label, single arm, multicentre study (study SG035-0004) in 58 patients with relapsed or refractory sALCL. See Table 24 for a summary of baseline patient and disease characteristics.
Response to treatment with Adcetris was assessed by IRF using the Revised Response Criteria for Malignant Lymphoma (Cheson, 2007). Treatment response was assessed by spiral CT of chest, neck, abdomen and pelvis; PET scans and clinical data. Response assessments were performed at cycles 2, 4, 7, 10, 13 and 16 with PET at cycles 4 and 7.
The ORR per IRF assessment was 86% (50 of 58 patients in the ITT set). CR was 59% (34 of 58 patients in the ITT set) and tumour reduction (of any degree) was achieved in 97% of patients. The estimated overall survival at 5 years was 60% (95% CI [47%, 73%]). The median observation time (time to death or last contact) from first dose was 71.4 months). The investigator assessments were generally consistent with the independent review of the scans. Of the patients treated, 9 responding patients went on to receive an allogeneic stem cell transplant and 9 responding patients went on to ASCT. For further efficacy results, see Table 25.
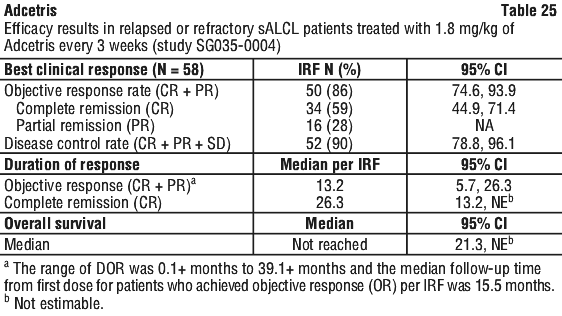
An exploratory intra-patient analysis showed that approximately 69% of the sALCL patients treated with Adcetris as part of the SG035-0004 clinical study experienced an improvement in clinical benefit as measured by longer progression free survival (PFS) compared with their most recent prior line of therapy.
Of the 17 patients (29%) who had B symptoms at baseline, 14 patients (82%) experienced resolution of all B symptoms in a median time from initiation of Adcetris of 0.7 months.
Study SGN35-006 (retreatment study). The efficacy of retreatment in patients who had previously responded (CR or PR) to treatment with Adcetris was evaluated in a phase 2, open-label, multicentre trial. Seven patients with relapsed sALCL received a starting dose of 1.8 mg/kg and one patient received a starting dose of 1.2 mg/kg of Adcetris administered intravenously over 30 minutes every 3 weeks. The median number of cycles was 8.5 (range, 2 to 30 cycles). Of the 8 sALCL patients, 3 were retreated twice for a total of 11 retreatment experiences. Retreatment with Adcetris resulted in 6 CRs (55%) and 4 PRs (36%), for an ORR of 91%. The median duration of response was 8.8 and 12.3 months in patients who achieved OR (CR+PR) and CR, respectively.
Cutaneous T-cell lymphoma (CTCL). Cutaneous T-cell lymphoma (study C25001). The efficacy and safety of Adcetris as a single agent was evaluated in a pivotal phase 3, open-label, randomised, multicentre study in 128 patients with histologically confirmed CD30+ CTCL. Patients were stratified by disease subtype (mycosis fungoides [MF] or primary cutaneous anaplastic large cell lymphoma [pcALCL]) and randomised 1:1 to receive either Adcetris or the physician's choice of either methotrexate or bexarotene. Patients with pcALCL received either prior radiation therapy or at least 1 prior systemic therapy and patients with MF received at least 1 prior systemic therapy. Patients were treated with 1.8 mg/kg of Adcetris intravenously over 30 minutes every 3 weeks for up to 16 cycles or physician's choice for up to 48 weeks. The median number of cycles was approximately 12 cycles in the Adcetris arm. In the physician's choice arm, the median duration of treatment (number of cycles) for patients receiving bexarotene was approximately 16 weeks (5.5 cycles) and 11 weeks (3 cycles) for patients receiving methotrexate. Table 26 provides a summary of the baseline patient and disease characteristics.

Patient reported skin symptom burden was assessed using the symptom domain of Skindex-29 quality of life questionnaire. Symptom burden reduction from baseline was observed in both groups across the study duration, however significantly greater symptom burden reduction based on mean maximum reduction from baseline was observed in the Adcetris arm compared to the physician's choice arm. The difference between the treatment arms for the maximum reduction from baseline (-18.9) exceeded all the estimated minimal important difference (MID) thresholds, demonstrating a clinically meaningful response.
5.2 Pharmacokinetic Properties
The pharmacokinetics of brentuximab vedotin were evaluated in phase 1 studies and in a population pharmacokinetic analysis of data from 314 patients. In all clinical trials, brentuximab vedotin was administered as an intravenous infusion.
Absorption. Monotherapy. Maximum concentrations of brentuximab vedotin ADC were typically observed at the end of infusion or the sampling time point closest to the end of infusion. A multiexponential decline in ADC serum concentrations was observed with a terminal half-life of approximately 4 to 6 days. Exposures were approximately dose proportional. Minimal to no accumulation of ADC was observed with multiple doses at every 3-week schedule, consistent with the terminal half-life estimate. Typical Cmax and AUC of ADC after a single 1.8 mg/kg in a phase 1 study was approximately 31.98 microgram/mL and 79.41 microgram/mL x day respectively.
MMAE is the major metabolite of brentuximab vedotin. Median Cmax, AUC and Tmax of MMAE after a single 1.8 mg/kg of the ADC in a phase 1 study was approximately 4.97 nanogram/mL, 37.03 nanogram/mL x day and 2.09 days respectively. MMAE exposures decreased after multiple doses of brentuximab vedotin with approximately 50% to 80% of the exposure of the first dose being observed at subsequent doses. In the first cycle, higher MMAE exposure was associated with an absolute decrease in neutrophil count.
Combination therapy. The pharmacokinetics of Adcetris in combination with AVD were evaluated in a single phase 3 study in 661 patients. Population pharmacokinetic analysis indicated that the pharmacokinetics of Adcetris in combination with AVD were consistent to that in monotherapy.
After multiple-dose, IV infusion of 1.2 mg/kg brentuximab vedotin every two weeks, maximal serum concentrations of ADC were observed near the end of the infusion and elimination exhibited a multi-exponential decline with a t1/2z of approximately 4 to 5 days. Maximal plasma concentrations of MMAE were observed approximately 2 days after the end of infusion, and exhibited a monoexponential decline with a t1/2z of approximately 3 to 4 days.
After multiple-dose, IV infusion of 1.2 mg/kg brentuximab vedotin every two weeks, steady-state trough concentrations of ADC and MMAE were achieved by cycle 3. Once steady-state was achieved, the PK of ADC did not appear to change with time. ADC accumulation (as assessed by AUC14D between cycle 1 and cycle 3) was 1.27-fold. The exposure of MMAE (as assessed by AUC14D between cycle 1 and cycle 3) appeared to decrease with time by approximately 50%.
The pharmacokinetics of Adcetris in combination with CHP were evaluated in a single phase 3 study in 223 patients (SGN35-014). After multiple-dose IV infusion of 1.8 mg/kg Adcetris every 3 weeks, the pharmacokinetics of ADC and MMAE were similar to those of monotherapy.
The pharmacokinetics of Adcetris was not studied in the BrECADD regimen.
Distribution. In vitro, the binding of MMAE to human serum plasma proteins ranged from 68-82%. MMAE is not likely to displace or to be displaced by highly protein bound medicines. In vitro, MMAE was a substrate of P-gp and was not an inhibitor of P-gp at clinical concentrations. In humans, the mean steady-state volume of distribution was approximately 6-10 L for ADC. Based on population PK estimation the typical apparent volume of distribution of MMAE in the central compartment was 7.37 L and the typical apparent volume of distribution of MMAE in the peripheral compartment was 36.4 L.
Metabolism. The antibody component of the ADC is expected to be catabolised as a protein with component amino acids recycled or eliminated. In vivo data in animals and humans suggest that only a small fraction of MMAE released from brentuximab vedotin is metabolised. The levels of MMAE metabolites have not been measured in human plasma. At least one metabolite of MMAE has been shown to be active in vitro. MMAE is a substrate of CYP3A4 and possibly CYP2D6. In vitro data indicate that the MMAE metabolism that occurs is primarily via oxidation by CYP3A4/5. In vitro studies using human liver microsomes indicate that MMAE inhibits only CYP3A4/5 at concentrations much higher than was achieved during clinical application. MMAE does not inhibit other isoforms. MMAE did not induce any major CYP450 enzymes in primary cultures of human hepatocytes.
Excretion. The ADC is eliminated by catabolism with a typical estimated CL and half-life of 1.457 L/day and 4-6 days respectively. The elimination of MMAE was limited by its rate of release from ADC, with a typical apparent CL and half-life of MMAE of 19.99 L/day and 3-4 days respectively.
An excretion study was undertaken in patients who received a dose of 1.8 mg/kg of brentuximab vedotin. Approximately 24% of the total MMAE administered as part of the ADC during a brentuximab vedotin infusion was recovered in both urine and faeces over a 1 week period. Of the recovered MMAE, approximately 72% was recovered in the faeces. A lesser amount of MMAE (28%) was excreted in the urine.
Pharmacokinetics in special populations. Population PK analysis showed that baseline serum albumin concentration was a significant covariate of MMAE clearance. The analysis indicated that MMAE clearance was 2-fold lower in patients with low serum albumin concentrations < 3.0 g/dL compared with patients with serum albumin concentrations within the normal range.
Hepatic impairment. The liver is a major route of elimination of the unchanged active metabolite MMAE.
A study evaluated the PK of brentuximab vedotin and MMAE after the administration of 1.2 mg/kg of Adcetris to patients with mild (Child-Pugh A; n = 1), moderate (Child-Pugh B; n = 5) and severe (Child-Pugh C; n = 1) hepatic impairment. Compared to patients with normal hepatic function, MMAE exposure increased approximately 2.3-fold in patients with hepatic impairment (see Section 4.2 Dose and Method of Administration).
Renal impairment. The kidney is a route of excretion of the unchanged active metabolite MMAE.
A study evaluated the PK of brentuximab vedotin and MMAE after the administration of 1.2 mg/kg of Adcetris to patients with mild (n = 4), moderate (n = 3) and severe (n = 3) renal impairment. Compared to patients with normal renal function, MMAE exposure increased approximately 1.9-fold in patients with severe renal impairment (creatinine clearance < 30 mL/min) (see Section 4.2 Dose and Method of Administration).
Elderly patients. The population pharmacokinetics of brentuximab vedotin were examined from several studies, including data from 380 patients up to 87 years old. The influence of age on pharmacokinetics was investigated and it was not a significant covariate. The safety profile in elderly patients with CTCL was consistent with that of younger patients, therefore no dosage adjustment is recommended for patients aged 65 and older.
Paediatric population. Clinical studies of brentuximab vedotin did not include sufficient numbers of patients below 18 years of age to determine whether the PK profile differs from adult patients.
5.3 Preclinical Safety Data
Genotoxicity. MMAE was genotoxic in the rat bone marrow micronucleus study through an aneugenic mechanism. This effect is consistent with the pharmacological effect of MMAE as a microtubule disrupting agent. MMAE was not mutagenic in the bacterial reverse mutation assay (Ames test) or the L5178Y mouse lymphoma forward mutation assay.
Carcinogenicity. Carcinogenicity studies with brentuximab vedotin or MMAE have not been conducted.
6 Pharmaceutical Particulars
6.1 List of Excipients
The reconstituted product contains trehalose dihydrate, sodium citrate dihydrate, citric acid monohydrate, and polysorbate 80 and water for injection.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
After reconstitution. After reconstitution/ dilution, from a microbiological point of view, the product should be used immediately. However, chemical and physical in-use stability has been demonstrated for 24 hours at 2°C-8°C.
6.4 Special Precautions for Storage
Store Adcetris at 2°C-8°C. Refrigerate. Do not freeze. Keep the vial in the original carton in order to protect from light. Do not use beyond the expiry date included on the carton/vial.
6.5 Nature and Contents of Container
Adcetris is supplied in a glass vial with a butyl rubber stopper and an aluminium/plastic flip-off seal, containing 50 mg Adcetris as a white to off-white lyophilized cake or powder. Each pack of Adcetris contains 1 vial.
6.6 Special Precautions for Disposal
Adcetris is for single use in one patient only. Any unused product or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
Chemical structure. Adcetris (brentuximab vedotin) is a CD30-directed antibody-drug conjugate (ADC) consisting of three components: 1) the chimeric IgG1 antibody cAC10, specific for human CD30, 2) the microtubule disrupting agent MMAE, and 3) a protease-cleavable linker that covalently attaches MMAE to cAC10.
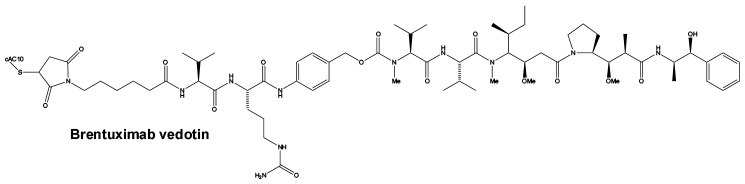
CAS number. 914088-09-8.
7 Medicine Schedule (Poisons Standard)
Prescription Only Medicine (Schedule 4).
Date of First Approval
20 December 2013
Date of Revision
17 December 2025
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.