Anagrelide Lupin
Brand Information
| Brand name | Anagrelide Lupin |
| Active ingredient | Anagrelide |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Anagrelide Lupin.
Summary CMI
Anagrelide Lupin
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I using Anagrelide Lupin?
Anagrelide Lupin contains the active ingredient anagrelide (as hydrochloride monohydrate). Anagrelide Lupin is used to treat essential thrombocythaemia.
For more information, see Section 1. Why am I using Anagrelide Lupin? in the full CMI.
2. What should I know before I use Anagrelide Lupin?
Do not use if you have ever had an allergic reaction to anagrelide (as hydrochloride monohydrate) or any of the ingredients listed at the end of the CMI.
Talk to your doctor or pharmacist if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use Anagrelide Lupin? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with Anagrelide Lupin and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use Anagrelide Lupin?
- The recommended adult starting dose is 1 mg/day, which can be taken orally in two divided doses.
- Take the tablet the same time each day.
More instructions can be found in Section 4. How do I use Anagrelide Lupin? in the full CMI.
5. What should I know while using Anagrelide Lupin?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using Anagrelide Lupin? in the full CMI.
6. Are there any side effects?
Serious side effects include: heart problems; lung problems; yellowing of the skin and eyes caused by inflammation of the liver (hepatitis); and severe abdominal or back pain (pancreatitis).
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
Anagrelide Lupin
Active ingredient(s): anagrelide (as hydrochloride monohydrate)
Consumer Medicine Information (CMI)
This leaflet provides important information about using Anagrelide Lupin. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using Anagrelide Lupin.
Where to find information in this leaflet:
1. Why am I using Anagrelide Lupin?
2. What should I know before I use Anagrelide Lupin?
3. What if I am taking other medicines?
4. How do I use Anagrelide Lupin?
5. What should I know while using Anagrelide Lupin?
6. Are there any side effects?
7. Product details
1. Why am I using Anagrelide Lupin?
Anagrelide Lupin contains the active ingredient anagrelide (as hydrochloride monohydrate). Anagrelide Lupin acts upon the bone marrow and prevents it from producing too many of the blood cells known as “platelets”.
Anagrelide Lupin is used to prevent too many platelets from being made. In a disease such as “thrombocythaemia”, the bone marrow produces too many of these cells, and the very large numbers of platelets in the blood can cause serious problems with blood circulation. By preventing too many platelets from being made, Anagrelide Lupin can help prevent these problems.
Your doctor may have prescribed Anagrelide Lupin for another reason. Ask your doctor if you have any questions about why Anagrelide Lupin has been prescribed for you.
Anagrelide Lupin is not addictive.
2. What should I know before I use Anagrelide Lupin?
Warnings
Do not use Anagrelide Lupin if:
- you are allergic to anagrelide, or any of the ingredients listed at the end of this leaflet.
Anagrelide Lupin contains lactose. These may cause a problem in a small number of people who are sensitive to them.
Always check the ingredients to make sure you can use this medicine. - you have severe liver problems.
- you are pregnant or breast feeding.
- the expiry date (EXP) printed on the bottle has passed.
If you take Anagrelide Lupin after the expiry date has passed, it may not work as well. - the packaging is torn or shows signs of tampering.
Check with your doctor or pharmacist if you:
- have any allergies;
- have, or have had, any liver or kidney disease;
- have heart disease, heart failure or are high risk of vascular events (thrombosis or bleeding);
- have an intolerance to some sugars, as Anagrelide Lupin contains sugars as lactose.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
If you become pregnant whilst taking Anagrelide Lupin, you should stop taking the capsules and see your doctor immediately.
Women taking Anagrelide Lupin and who are at risk of becoming pregnant should make sure that they are using adequate contraception.
Use in children
Anagrelide Lupin should not be taken by anyone under 16 years of age.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and Anagrelide Lupin may interfere with each other. These include:
- aspirin or a medicine containing aspirin;
- medicines used to treat depression, such as fluvoxamine;
- medicines used to treat gastrointestinal problems, such as omeprazole;
- medicines used to treat severe asthma and breathing problems, such as theophylline;
- medicines used to treat heart disorders, such as milrinone;
- other medicines used to treat conditions affecting the platelets in your blood;
- medicines containing sucralfate.
Your doctor may have to change your dose of other medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while using this medicine.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect Anagrelide Lupin.
4. How do I use Anagrelide Lupin?
How much to take / use
- The recommended adult starting dose of anagrelide is 1 mg/day, which can be taken orally in two divided doses. This dosage will then be adjusted until your doctor has decided which dosage is best for you.
- Any single dose taken during the day should not exceed 2.5 mg. Your total daily dose should not exceed four times this, ie. 10 mg (20 of the 0.5 mg capsules).
- If you are elderly, you should take the normal adult dose.
- If you are not sure, ask your doctor or pharmacist for advice.
- Follow all the directions given to you by your doctor or pharmacist carefully. They may differ from the information in this leaflet.
When to take Anagrelide Lupin
- Take Anagrelide Lupin at the same time each day. This will help you remember when to take the capsules.
- Food reduces the absorption of Anagrelide Lupin slightly, but this does not have any effect on the ability to reduce your platelet count.
How long to take Anagrelide Lupin
- You should not normally stop taking Anagrelide Lupin unless your doctor tells you to. If Anagrelide Lupin has been successfully reducing the excess of platelets in your blood, stopping Anagrelide Lupin will cause the number of platelets in your blood to rise again within three or four days, so that the risk of problems with blood circulation may return.
- Abrupt treatment discontinuation or substantial reduction in Anagrelide Lupin's dosage should be avoided, due to potentially severe clotting complications.
- If you feel unwell during your course of treatment, tell your doctor.
If you forget to take Anagrelide Lupin
If you forget to take a dose of Anagrelide Lupin, skip that dose completely. Take your next dose at the normal time it is due.
Do not take a double dose to make up for the dose you missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take Anagrelide Lupin, ask your pharmacist for some hints.
If you use too much Anagrelide Lupin
If you think that you have used too much Anagrelide Lupin, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26); or - contact your doctor; or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
Symptoms of an overdose may include fast heartbeat, vomiting and bleeding.
5. What should I know while using Anagrelide Lupin?
Things you should do
- Make sure that all your doctors and pharmacists know about your use of Anagrelide Lupin. Remind them if any new medicines are about to be started.
- Tell your doctor immediately if you experience shortness of breath and fatigue while taking Anagrelide Lupin.
Remind any doctor, dentist or pharmacist you visit that you are using Anagrelide Lupin.
Things you should not do
- Do not take Anagrelide Lupin to treat any complaint other than that directed by your doctor. It may not be safe to take Anagrelide Lupin for another complaint.
- Do not give your medicine to anyone else, even if they have the same condition as you. It may not be safe for another person to take Anagrelide Lupin.
- Do not stop taking your Anagrelide Lupin or change the dosage without checking with your doctor.
Driving or using machines
Anagrelide Lupin may cause dizziness in some patients. Be careful before you drive or use any machines or tools until you know how Anagrelide Lupin affects you.
Looking after your medicine
- Keep Anagrelide Lupin capsules in their bottle until it is time to take your dose.
- Keep Anagrelide Lupin in a cool dry place where the temperature stays below 25°C.
Follow the instructions on the carton on how to take care of your medicine properly.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink; or
- in the car or on window sills.
Keep it where young children cannot reach it.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions.
Less serious side effects
| Less serious side effects | What to do |
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
As a precaution, your doctor may have your blood, liver and kidney tested regularly during treatment with Anagrelide Lupin.
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor' prescription.
What Anagrelide Lupin contains
| Active ingredient (main ingredient) | Anagrelide (as hydrochloride monohydrate) |
| Other ingredients (inactive ingredients) | Lactose monohydrate Microcrystalline cellulose Povidone Croscarmellose sodium Magnesium stearate Anhydrous lactose The capsule shell contains:
|
| Potential allergens | Lactose |
Do not take this medicine if you are allergic to any of these ingredients.
What Anagrelide Lupin looks like
Anagrelide Lupin is a capsule with an opaque white body and cap (AUST R 276297). The capsule is filled with white to off-white powder.
Who distributes Anagrelide Lupin
Generic Health Pty Ltd
Suite 2, Level 2
19-23 Prospect Street
Box Hill, VIC, 3128
Australia

 +61 3 9809 7900
+61 3 9809 7900

This leaflet was prepared in September 2025.
Brand Information
| Brand name | Anagrelide Lupin |
| Active ingredient | Anagrelide |
| Schedule | S4 |
MIMS Revision Date: 01 April 2025
Notes
Distributed by Generic Health Pty Ltd
1 Name of Medicine
Anagrelide hydrochloride.
2 Qualitative and Quantitative Composition
Anagrelide capsules contain the active ingredient anagrelide (as hydrochloride monohydrate) equivalent to 0.5 mg of anagrelide.
Excipients with known effect. Lactose and lactose monohydrate. Contains sugars as lactose.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Anagrelide Lupin is a capsule with an opaque white body and cap.
Each Anagrelide Lupin capsules contain 0.5 mg of anagrelide as the active ingredient.
4 Clinical Particulars
4.1 Therapeutic Indications
Anagrelide Lupin capsules are indicated for the treatment of essential thrombocythaemia.
4.2 Dose and Method of Administration
Treatment with Anagrelide Lupin capsules should be initiated under close medical supervision. The recommended starting dosage of Anagrelide Lupin for adult patients is 1 mg/day, which should be administered orally in two divided doses (0.5 mg/dose). The starting dose should be maintained for at least a week. The dose should then be adjusted to the lowest effective dose required to reduce and maintain platelet count below 600 x 109/L and ideally at levels between 150-400 x 109/L. The dose should be increased by not more than 0.5 mg/day in any one week.
Dosage should not exceed 10 mg/day or 2.5 mg in a single dose because of the hypotensive effect of anagrelide (see Section 5.1 Pharmacodynamic Properties). The decision to treat asymptomatic young adults with essential thrombocythaemia should be individualised (see Section 4.4 Special Warnings and Precautions for Use).
Hepatic impairment. For patients with moderate hepatic impairment, the recommended starting dose is 0.5 mg/day, to be maintained for a minimum of one week with close monitoring of cardiovascular effects. Anagrelide Lupin is not recommended for patients with severe hepatic impairment.
Patient monitoring. To monitor the effect of anagrelide and prevent the occurrence of thrombocytopenia, platelet counts should be performed every two days during the first week of treatment and at least weekly thereafter until the maintenance dosage is reached.
Typically, platelet count begins to respond within 7 to 14 days at the proper dosage. Most patients will experience an adequate response at a dose of 1.5 to 3.0 mg/day. Patients with known or suspected heart disease, renal insufficiency, or hepatic dysfunction should be monitored closely. Where immediate reduction of the platelet count is required, pheresis may be a more appropriate therapeutic intervention (see Section 4.4 Special Warnings and Precautions for Use).
4.3 Contraindications
Anagrelide Lupin is contraindicated in patients who have developed hypersensitivity to anagrelide hydrochloride or any of its excipients (see Section 6.1 List of Excipients) and in patients with severe hepatic impairment. Exposure to anagrelide is increased 8-fold in patients with moderate hepatic impairment. Use of anagrelide in patients with severe hepatic impairment has not been studied.
4.4 Special Warnings and Precautions for Use
Patients with rare hereditary problems of galactose intolerance, the Lapp lactase deficiency or glucose-galactose malabsorption should not take Anagrelide Lupin as it contains lactose.
Cardiovascular. Therapeutic doses of anagrelide may cause cardiovascular effects, including vasodilation, tachycardia, palpitations and congestive cardiac failure. A pre-treatment cardiovascular examination (including investigations such as echocardiography, electrocardiogram) is recommended for all patients. Patients should be monitored during treatment of cardiovascular effects and further investigations carried out as necessary. Hypokalaemia or hypomagnesaemia must be corrected prior to anagrelide administration. Anagrelide should be used with caution in patients with known or suspected heart disease or at high risk of vascular events, and only if the potential benefits of therapy outweigh the potential risks (also see Patient monitoring/ effects on laboratory tests in this section).
Anagrelide has been shown to increase both the heart rate and QTc interval in healthy volunteers. The clinical impact of this effect is unknown (see Section 5.1 Pharmacodynamic Properties).
Caution should be taken when using anagrelide in patients with known risk factors for prolongation of the QT interval, such as congenital long QT syndrome, a known history of acquired QTc prolongation, medicinal products that can prolong QTc interval and hypokalaemia.
Care should also be taken in populations that may have a higher maximum plasma concentration (Cmax) of anagrelide or its active metabolite, 3-hydroxy-anagrelide, e.g. hepatic impairment or use with CYP1A2 inhibitors (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Pulmonary hypertension. Cases of pulmonary hypertension have been reported in patients treated with anagrelide. Patients should be evaluated for signs and symptoms of underlying cardiopulmonary disease prior to initiating and during anagrelide therapy.
Bleeding. Use of concomitant anagrelide and aspirin (acetylsalicylic acid) has been associated with major haemorrhagic events (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Use in renal impairment. Patients with renal impairment (serum creatinine ≥ 0.18 mmol/L) should be monitored closely since they are at greater risk of renal toxicity while receiving anagrelide (see Section 4.8 Adverse Effects (Undesirable Effects)).
Use in hepatic impairment. Exposure to anagrelide is increased 8-fold in patients with moderate hepatic impairment. Use of anagrelide in patients with severe hepatic impairment has not been studied. The potential risks and benefits of anagrelide therapy in a patient with mild or moderate impairment of hepatic function should be assessed before and during treatment being commenced. In patients with moderate hepatic impairment, dose reduction is required, and patients should be carefully monitored for cardiovascular effects (see Section 4.2 Dose and Method of Administration for specific dosing recommendations).
Patients with hepatic impairment (serum bilirubin, AST or other measures of hepatic function > 1.5 times the upper limit of normal) should be monitored closely while receiving anagrelide since anagrelide may worsen hepatic impairment (see Section 4.8 Adverse Effects (Undesirable Effects), Hepatobiliary disorders).
Patient monitoring/ effects on laboratory tests. Anagrelide therapy requires close clinical supervision of the patient. While the platelet count is being lowered (usually during the first two weeks of treatment), platelet count should be performed every 2 days during the first week of treatment and at least weekly thereafter until the maintenance dosage is reached (see Section 4.2 Dose and Method of Administration). As cases of hepatitis have been reported from post-marketing surveillance, it is recommended that liver function tests (ALT and AST) are performed before anagrelide treatment is initiated and at regular intervals thereafter. A full blood count (haemoglobin, white blood cells and platelets), renal function (serum creatinine, urea) tests and electrolytes (potassium, magnesium and calcium) should continue to be monitored during anagrelide therapy. Patients should be regularly assessed during anagrelide therapy for the emergence of cardiovascular effects which may require further examination and investigation. The QTc interval should be closely monitored during anagrelide treatment.
Paediatric use. The efficacy and safety of anagrelide in patients under the age of 16 years have not been established.
Use in the elderly. The observed pharmacokinetic differences between elderly and young patients with essential thrombocythaemia (ET) do not warrant using a different starting regimen or different dose titration step to achieve an individual patient-optimised anagrelide regimen.
Cessation of Anagrelide Lupin treatment. In general, interruption of anagrelide treatment is followed by an increase in platelet count. After sudden stoppage of anagrelide therapy, the increase in platelet count can be observed within four days. It should be noted that there is risk of thromboembolic events during this rebound phase. The dynamics of the rise in platelet count following interruption of therapy have been studied in only a very small number of patients, and data from normal controls suggests that a rebound to beyond pretreatment levels occurs in some individuals.
Therefore, platelet count should be monitored frequently.
Thrombotic risk. Abrupt treatment discontinuation or substantial reduction of anagrelide's dose should be avoided due to the risk of sudden increase in platelet counts, which may lead to potentially fatal thrombotic complications, such as cerebral infarction (see Section 4.2 Dose and Method of Administration). Patients should be advised how to recognise early signs and symptoms suggestive of thrombotic complications, such as cerebral infarction, and if symptoms occur to seek medical assistance.
Treatment discontinuation. In the event of dosage interruption or treatment withdrawal, platelets should be monitored frequently (see Section 4.2 Dose and Method of Administration).
4.5 Interactions with Other Medicines and Other Forms of Interactions
Limited PK and/or PD studies investigating possible interactions between anagrelide and other medicinal products have been conducted.
Warfarin or digoxin. In vivo interaction studies in humans have demonstrated that digoxin and warfarin do not affect the PK properties of anagrelide, nor does anagrelide affect the PK properties of digoxin or warfarin.
Aspirin (acetylsalicylic acid) and drugs that increase bleeding risk. At therapeutic doses, anagrelide may potentiate the effects of other medicinal products that inhibit platelet aggregation. In two clinical interaction studies in healthy subjects, co-administration of single-dose anagrelide 1 mg and aspirin 900 mg or repeat-dose anagrelide 1 mg once daily and aspirin 75 mg once daily showed greater anti-platelet aggregation effects than administration of aspirin alone. Co-administered anagrelide 1 mg and aspirin 900 mg single-doses had no effect on bleeding time, prothrombin time (PT) or activated partial thromboplastin time (aPTT). In the repeat-dose study, there was a short-lived decrease in ex vivo collagen-induced platelet aggregation beyond the effects of aspirin alone for the first 2 hours after administration. In some ET patients concomitantly treated with aspirin and anagrelide, major haemorrhages have occurred. The potential risks and benefits of concomitant use of anagrelide with aspirin should be assessed, particularly in patients with a high-risk profile for haemorrhage, before treatment is commenced.
Other PDE3 inhibitors. Anagrelide is an inhibitor of cyclic AMP phosphodiesterase III (PDE3). The effects of drugs with similar properties such as inotropes (e.g. milrinone) may be exacerbated by anagrelide.
CYP1A2 inhibitors. Anagrelide is primarily metabolised by CYP1A2. It is known that CYP1A2 is inhibited by several medicinal products, including fluvoxamine and ciprofloxacin, and such medicinal products could theoretically adversely influence the clearance of anagrelide, thereby increasing plasma concentrations. Anagrelide demonstrated inhibitory activity towards CYP1A2 in vitro which presents a theoretical potential for interaction with other co-administered medicinal products sharing that clearance mechanism e.g. theophylline.
CYP1A2 inducers. CYP1A2 inducers could decrease the exposure of anagrelide. Patients taking concomitant CYP1A2 inducers (e.g. omeprazole) may need to have their dose titrated to compensate for the decrease in anagrelide exposure.
Preclinical data indicate an augmented anticoagulant effect when heparin and anagrelide were used in combination.
There is a single case report which suggests that sucralfate may interfere with anagrelide absorption.
Food has no clinically significant effect on the bioavailability of anagrelide.
Although additional drug interaction studies have not been conducted, the most common medications used concomitantly with anagrelide in clinical trials have been aspirin, paracetamol, furosemide, iron, ranitidine, hydroxyurea, and allopurinol. The most frequently used concomitant cardiac medication has been digoxin. Other than aspirin, there is no clinical evidence to suggest that anagrelide interacts with any of these compounds.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. Studies in animals have shown reproductive toxicity. Anagrelide hydrochloride at oral doses up to 240 mg/kg/day (1,440 mg/m2/day, 195 times the maximum recommended human dose based on body surface area) had no effect on fertility of male rats. However, in female rats, given oral doses of 60 mg/kg/day (360 mg/m2/day, 49 times the maximum recommended human dose based on body surface area) or higher, it disrupted implantation when administered in early pregnancy. A no-effect dose level was not established.
Use in pregnancy. (Category B31)
1 Drugs which have been taken by only a limited number of pregnant women and women of childbearing age, without an increase in the frequency of malformation or other direct or indirect harmful effects on the human foetus having been observed. Studies in animals have shown evidence of an increased occurrence of foetal damage, the significance of which is considered uncertain in humans.
Anagrelide is not recommended in women who are or may become pregnant. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential harm to the foetus. Women of child-bearing potential should be instructed that they must not be pregnant and that they should use contraception while taking anagrelide. Anagrelide may cause foetal harm when administered to a pregnant woman. There are no adequate studies of anagrelide use in pregnant women.
Five women became pregnant while on anagrelide treatment at doses of 1 to 4 mg/day. Treatment was stopped as soon as it was realised they were pregnant. All delivered normal, healthy babies.
No teratogenic effects were observed in pregnant rats at oral doses up to 900 mg/kg/day (5400 mg/m2/day, 730 times the maximum recommended human dose based on body surface area), or in pregnant rabbits at oral doses up to 20 mg/kg/day (240 mg/m2/day, 32 times the maximum recommended human dose based on body surface area). However, increased embryonic deaths and suppression of foetal growth were seen in rats at oral doses of 60 mg/kg/day (360 mg/m2/day, 49 times the maximum recommended human dose based on body surface area) or greater, and ossification was retarded at 100 mg/kg/day or greater. When administered to rats in late pregnancy at oral doses of 60 mg/kg/day or higher, it retarded or blocked parturition, resulting in maternal and neonatal deaths.
Use in lactation. It is not known whether this drug is excreted in human milk. When administered to lactating rats, anagrelide hydrochloride at doses greater than 60 mg/kg/day (360 mg/m2/day, 49 times the maximum recommended human dose based on body surface area) decreased survival of the offspring. Because many drugs are excreted in human milk and in view of the unknown risks of anagrelide to the infant, a decision to discontinue nursing or to discontinue the drug should be seriously considered, taking into account the importance of the drug to the mother.
4.7 Effects on Ability to Drive and Use Machines
Anagrelide may cause dizziness in some patients. Caution should be shown when driving or operating machinery whilst on treatment with anagrelide.
4.8 Adverse Effects (Undesirable Effects)
While most reported adverse events during anagrelide therapy have been mild in intensity and have decreased in frequency with continued therapy, serious adverse events reported in patients with ET and/or in patients with thrombocythaemias or other aetiologies include: congestive heart failure, myocardial infarction, cardiomyopathy, cardiomegaly, complete heart block, atrial fibrillation, torsades de pointes, ventricular tachycardia, cerebrovascular accident, pericarditis, pericardial effusion, pleural effusion, pulmonary infiltrates, pulmonary fibrosis, pulmonary hypertension, pancreatitis, hepatitis, gastric/duodenal ulceration, tubulointerstitial nephritis and seizure.
Of the 551 patients treated with anagrelide for a mean duration of 65 weeks, 82 (15%) were discontinued from the study because of adverse events or abnormal laboratory test results. The most common adverse events for treatment discontinuation were headache, diarrhoea, oedema, palpitation and abdominal pain. Overall, the occurrence rate of all adverse events was 17.9 per 1,000 treatment days. The occurrence rate of adverse events increased at higher dosages of anagrelide.
Adverse reactions arising from clinical studies, post-authorisation safety studies, and spontaneous reports are presented below. Within the system organ classes, they are listed under the following headings: very common (≥ 1/10); common (≥ 1/100 to <1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000), not known (cannot be estimated from the available data). (* Denotes post-marketing ADRs).
General disorders and administration site conditions. Very common: headache (44.5%), asthenia (22.1%), pain, other than abdominal, chest or back (14.7%).
Common: fever, influenza-like illness, chills, neck pain, photosensitivity, paraesthesia, back pain, malaise.
Uncommon: oedema, chest pain.
Cardiac and vascular disorders. Very common: palpitations (27.2%), oedema (19.8%).
Common: arrhythmia, haemorrhage, cardiovascular disease, cerebrovascular accident, angina pectoris, congestive heart failure, hypertension, orthostatic hypotension, vasodilatation, chest pain, tachycardia, peripheral oedema.
Uncommon: ventricular tachycardia, supraventricular tachycardia, atrial fibrillation.
Rare: myocardial infarction, cardiomyopathy, cardiomegaly, pericardial effusion.
Not known: *Torsades de pointes, Prinzmetal angina.
Gastrointestinal disorders. Very common: diarrhoea (24.3%), abdominal pain (17.4%), nausea (15.1%), flatulence (10.5%).
Common: constipation, GI distress, GI haemorrhage, gastritis, melena, aphthous stomatitis, eructation, nausea, vomiting, dyspepsia.
Uncommon: pancreatitis.
Blood and lymphatic system disorders. Common: anaemia, thrombocytopenia, ecchymosis, lymphadenoma.
Platelet counts below 100 x 109/L occurred in 35 patients and reduction below 50 x 109/L occurred in 7 of the 551 ET patients while on anagrelide therapy. Thrombocytopenia promptly recovered upon discontinuation of anagrelide.
Hepatobiliary disorders. Common: elevated liver enzymes.
Not known: *hepatitis.
Musculoskeletal and connective tissue disorders. Common: arthralgia, myalgia, leg cramps.
Uncommon: back pain.
Nervous system disorders. Very common: dizziness (14.5%), headache.
Common: depression, hypoesthesia, somnolence, confusion, insomnia, nervousness, amnesia, migraine, syncope.
Uncommon: paraesthesia.
Not known: cerebral infarction.
Psychiatric disorders. Uncommon: depression, confusional state, insomnia, nervousness.
Metabolism and nutritional disorders. Common: dehydration, decreased appetite.
Respiratory, thoracic and mediastinal disorders. Very common: dyspnoea (10.5%).
Common: rhinitis, epistaxis, respiratory disease, sinusitis, pneumonia, bronchitis, asthma, cough, pharyngitis.
Uncommon: Pleural effusion, pulmonary hypertension.
Rare: lung infiltration.
Not known: *interstitial lung disease including pneumonitis and allergic alveolitis.
Skin and subcutaneous tissue disorders. Common: pruritus, skin disease, alopecia, rash including urticaria.
Uncommon: ecchymosis.
Eye, ear and labyrinth disorders. Common: amblyopia, visual impairment, tinnitus, visual field abnormality, diplopia.
Renal and urinary disorders. Common: dysuria, haematuria.
Rare: renal failure.
Not known: *tubulointerstitial nephritis.
Investigations. Uncommon: hepatic enzymes increased.
* Denotes post-marketing ADRs.
Of the 551 patients, 10 were found to have renal abnormalities. Six of the 10 experienced renal failure (approximately 1%) while on anagrelide treatment; in two, the renal failure was considered to be possibly related to anagrelide treatment. The remaining 4 were found to have pre-existing renal impairment and were successfully treated with anagrelide. Doses ranged from 1.5-6.0 mg/day, with exposure periods of 2 to 12 months. Serum creatinine remained within normal limits and no dose adjustment was required because of renal insufficiency.
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medical product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
4.9 Overdose
Acute toxicity and symptoms. Symptoms of acute toxicity were decreased motor activity in mice and rats and softened stools and decreased appetite in monkeys.
At higher than recommended doses, anagrelide has been shown to cause reductions in blood pressure, with occasional hypotension. There have been a small number of post-marketing case reports of intentional overdose with anagrelide. Reported symptoms include sinus tachycardia and vomiting. Symptoms resolved with conservative management. Platelet reduction from anagrelide therapy is dose-related; therefore thrombocytopenia, which can potentially cause bleeding, is expected from overdosage. Should overdosage occur, cardiac, and central nervous system toxicity can also be expected.
Management and treatment. In case of overdosage, close clinical supervision of the patient is required; this especially includes monitoring of the platelet count for thrombocytopenia. Dosage should be decreased or stopped, as appropriate, until the platelet count returns within the normal range.
For information on the management of overdose, contact the Poison Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Healthy male volunteers given anagrelide demonstrated dose-related reductions in platelet counts. The reduction after 8-10 days treatment with anagrelide 0.5 mg bd was 24-30% and with 1 mg bd, 30-44%. After 1 mg mane for 30 days, the average reduction in platelet count was 15%. Platelet counts returned to normal within one week of ceasing treatment.
Mechanism of action. The precise mechanism by which anagrelide reduces blood platelet count is unknown. In cell culture studies, anagrelide suppressed expression of transcription factors including GATA-1 and FOG-1 required for megakaryocytopoiesis, ultimately leading to reduced platelet production.
Pharmacodynamic effects. In vitro studies of the growth of human megakaryocyte colonies in tissue culture showed that anagrelide disrupted the postmitotic phase of megakaryocyte development reducing megakaryocyte size and ploidy. Anagrelide did not have a thrombocytopenic effect in the animal models tested, rats, dogs and monkeys, at doses ≤ 10 mg/kg/day.
Anagrelide at doses ≥ 1 mg inhibited ADP- and collagen-induced platelet aggregation in healthy volunteers. Anagrelide 0.5 mg twice daily for 14 days followed by anagrelide 1 mg twice daily for a further 14 days reduced haemoglobin concentration by a median 12 g/L. In in vitro studies of human blood, anagrelide inhibited cyclic AMP phosphodiesterase.
Anagrelide produces dose-dependent vasodilation, decreasing blood pressure and increasing heart rate and ventricular contractility. In a pharmacodynamic study, a dose of 5 mg caused orthostatic hypotension and dizziness in nine healthy volunteers (average fall in standing blood pressure = 22/15 mmHg). Only minimal changes in blood pressure were observed following a dose of 2 mg.
Effects on heart rate and QTc interval. The effect of two dose levels of anagrelide (0.5 mg and 2.5 mg single doses) on the heart rate and QTc interval was evaluated in a double-blind, randomised, placebo- and active-controlled, cross-over study in 60 healthy adult men and women.
A dose-related increase in heart rate was observed during the first 12 hours, with the maximum increase occurring around the time of maximal concentrations. The maximum change in mean heart rate occurred at 2 hours after administration and was +7.8 beats per minute (bpm) for 0.5 mg and +29.1 bpm for 2.5 mg.
An apparent transient increase in mean QTc was observed for both doses during periods of increasing heart rate and the maximum change in mean QTcF (Fridericia correction) was +5.0 msec (upper 2-sided 90% CI 8.0 msec) occurring at 2 hours for 0.5 mg and +10.0 msec (upper 2-sided 90% CI 12.7 msec) occurring at 1 hour for 2.5 mg. Anagrelide exposure was higher in women than men (Cmax 55-75% higher; AUC 90% higher) and women had higher heart rate changes (and QTc changes) than men around the time of Tmax.
Note. The recommended starting dosage of Anagrelide Lupin is 0.5 mg twice daily and should be increased by not more than 0.5 mg/day in any one week (see Section 4.2 Dose and Method of Administration).
Clinical trials. A total of 551 patients with Essential Thrombocythaemia (ET) were treated with anagrelide in two uncontrolled trials and in compassionate use. Patients with ET were diagnosed based on the following criteria: platelet count ≥ 900 x 109/L on two determinations; profound megakaryocytic hyperplasia in bone marrow; absence of Philadelphia chromosome; normal red cell mass; normal serum iron and ferritin, and normal marrow iron stores.
The mean duration of anagrelide therapy for study patients was 65 weeks; 23% of patients received treatment for 2 years. In the main trial, 274 ET patients were treated with anagrelide starting at doses of 0.5-2.0 mg every 6 hours. The dose was increased if the platelet count was still high, but to no more than 12 mg each day. The criteria for defining subjects as "responders" were reduction in platelets for at least 4 weeks to ≤ 600 x 109/L, or by at least 50% from baseline value. Subjects treated for less than 4 weeks were not considered evaluable. 79% of evaluable patients (n=254) responded and 73% of patients receiving at least one dose (n=274) responded. The reduction in mean platelet count is depicted graphically in Figure 1 and Table 1.
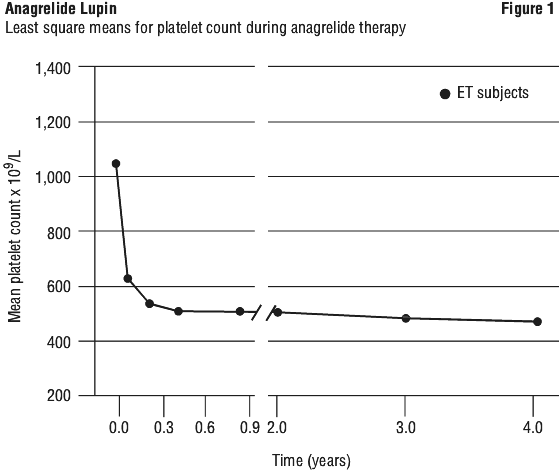
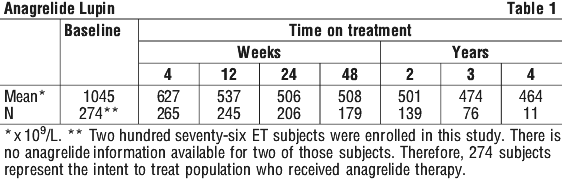
Both groups achieved similar control of platelet count within 9 months of trial entry.
Anagrelide + aspirin were associated with a significantly higher incidence of arterial thrombosis (9.1% vs 4.2%), serious haemorrhage (5.4% vs 2.0%) and transformation to myelofibrosis (4.0% vs 1.2%) in this high risk group compared to hydroxyurea + aspirin. Rates of death, from any cause, were not significantly different between the two groups.
5.2 Pharmacokinetic Properties
Absorption. Single oral doses of 1-2 mg anagrelide are absorbed rapidly in healthy male volunteers, mean Tmax being 0.9 h. Following administration of 14C-anagrelide in people, more than 70% of radioactivity was recovered in urine. Based on limited data, there appears to be a trend toward dose linearity between doses of 0.5 mg and 2.0 mg. At fasting and at a dose of 0.5 mg of anagrelide, the plasma half-life is 1.3 hours. The available plasma concentration time data at steady state in patients show that anagrelide does not accumulate in plasma after repeated administration. Anagrelide protein binding in human plasma is 91%.
Pharmacokinetic data obtained from healthy Caucasian volunteers comparing the pharmacokinetics of anagrelide in the fed and fasted states showed that administration of a 1 mg dose of anagrelide with food decreased the Cmax by 14%, but increased the AUC by 20%. Food also reduced the Cmax of 3-hydroxy anagrelide.
Metabolism. The drug is extensively metabolised; less than 1% is recovered in the urine as anagrelide. Two major metabolites have been identified RL603 and 3-hydroxy anagrelide. RL603 is considered pharmacologically inactive whilst 3-hydroxy anagrelide is pharmacologically active, being equipotent as the parent compound in terms of platelet inhibition and 40 times more potent as an inhibitor of phosphodiesterase III.
Excretion. The half-life of 3-hydroxy anagrelide is approximately 3 hours. Metabolites are excreted in urine (79%) and faeces (21%). Excretion is > 97% complete within 5 days. Anagrelide is metabolised by CYP1A2, and therefore there is potential for interaction with other co-administered drugs that alter (inhibit or induce) CYP1A2-mediated metabolism (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Special population pharmacokinetics. Pharmacokinetic (PK) data from paediatric (age range 7-14 years) and adult (age range 16-86 years) patients with thrombocythaemia secondary to a myeloproliferative disorder (MPD), indicate that dose and body weight-normalised exposure, Cmax and AUCt, of anagrelide were lower in the paediatric patients compared to the adult patients (Cmax 48%, AUCt 55%).
There were no apparent differences between patient groups (paediatric versus adult patients) for Tmax and t1/2 for anagrelide, 3-hydroxy anagrelide, or RL603.
Pharmacokinetic data from fasting elderly patients with ET (age range 65-75 years) compared to fasting adult patients (age range 22-50 years) indicate that the Cmax and AUC of anagrelide were 36% and 61% higher respectively in elderly patients, but that the Cmax and AUC of the active metabolite, 3-hydroxy anagrelide, were 42% and 37% lower respectively in the elderly patients. These differences were likely to be caused by lower presystemic metabolism of anagrelide to 3-hydroxy anagrelide in the elderly patients.
Renal impairment. A pharmacokinetic study at a single dose of 1 mg anagrelide in subjects with severe renal impairment (creatinine clearance < 30 mL/min) showed no significant effects on the pharmacokinetics of anagrelide. The pharmacokinetic results show that the exposure to 3-hydroxy anagrelide is higher (100% increase in plasma elimination half-life from 3 to 6 hours) in severe renally impaired patients although the Cmax did not differ.
Hepatic impairment. A pharmacokinetic study at a single dose of 1 mg anagrelide in subjects with moderate hepatic impairment showed an 8-fold increase in total exposure (AUC) to anagrelide.
5.3 Preclinical Safety Data
Genotoxicity. Anagrelide did not cause gene mutations in bacterial or mammalian cells, nor was it clastogenic in the human lymphocyte chromosome aberration test in vitro or the mouse micronucleus test in vivo.
Carcinogenicity. In a two year rat carcinogenicity study, a higher incidence of uterine adenocarcinoma was observed in females receiving 30 mg/kg/day (99 times human AUC for anagrelide and 18 times human AUC for metabolite 3-hydroxyanagrelide) with a NOEL of 10 mg/kg/day (6 times human AUC for anagrelide and twice human AUC exposure for metabolite 3-hydroxyanagrelide after the maximum recommended clinical dose of 10 mg/day). Adrenal phaeochromocytomas were increased in males receiving 3 mg/kg/day and above, and in females receiving 10 mg/kg/day and above. A NOEL was not established in males and for females was 3 mg/kg/day (1.6 times human AUC exposure to anagrelide and less than the human exposure to metabolite 3-hydroxyanagrelide after the maximum recommended clinical dose of 10 mg/day). Adrenal phaeochromocytomas were also found in a one year rat study.
No long-term data in humans are available to evaluate the carcinogenic potential of anagrelide hydrochloride. The maximum duration of human exposure in clinical trials was 4 years.
6 Pharmaceutical Particulars
6.1 List of Excipients
Anagrelide Lupin capsules contain the following inactive ingredients: lactose monohydrate, croscarmellose sodium, povidone, microcrystalline cellulose, magnesium stearate and anhydrous lactose.
The capsule shells consist of gelatin, titanium dioxide, iron oxide black.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 25°C.
6.5 Nature and Contents of Container
HDPE bottles with child resistant polypropylene closures containing 100 capsules.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
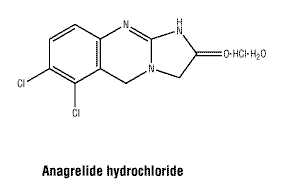
Chemical name: 6,7-dichloro-3,5-dihydroimidazo[2,1-b]quinazolin-2(1H)-one hydrochloride monohydrate.
Molecular formula: C10H7Cl2N3O.HCl.H2O.
Molecular weight: 310.56.
Anagrelide (as hydrochloride monohydrate) is an orally active quinazolin derivative.
Anagrelide hydrochloride monohydrate is a white to off-white powder. It is very slightly soluble in water, acetonitrile, slightly soluble in acetic acid and ethanol, sparingly soluble in methanol and soluble in formic acid solutions.
7 Medicine Schedule (Poisons Standard)
S4 (Prescription Only Medicine).
Date of First Approval
03 August 2017
Date of Revision
03 February 2025
Summary Table of Changes
Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.