ARX-Abiraterone
Brand Information
| Brand name | ARX-Abiraterone |
| Active ingredient | Abiraterone acetate |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the ARX-Abiraterone.
Summary CMI
ARX-Abiraterone
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I using ARX-Abiraterone?
ARX-Abiraterone contains the active ingredient abiraterone acetate. ARX-Abiraterone is used to treat prostate cancer that has spread to other parts of the body. It reduces the levels of the sex hormone testosterone.
For more information, see Section 1. Why am I using ARX-Abiraterone? in the full CMI.
2. What should I know before I use ARX-Abiraterone?
Do not use if you have ever had an allergic reaction to abiraterone acetate or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use ARX-Abiraterone? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with ARX-Abiraterone and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use ARX-Abiraterone?
- Always take ARX-Abiraterone exactly as your doctor has told you. You must check with your doctor if you are not sure.
- The usual daily dose of ARX-Abiraterone is four 250 mg tablets or two 500 mg tablets taken as a single dose.
More instructions can be found in Section 4. How do I use ARX-Abiraterone? in the full CMI.
5. What should I know while using ARX-Abiraterone?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine | 250 mg tablets: Store Below 25°C 500 mg tablets: Store below 30°C |
For more information, see Section 5. What should I know while using ARX-Abiraterone? in the full CMI.
6. Are there any side effects?
Serious side effects include chest pain, heartbeat disorders, heart failure, rapid heart rate, muscle pains, adrenal gland problems and anaphylactic reaction. The most common side effects of ARX-Abiraterone are diarrhoea, fluid in legs and feet, low blood potassium, urinary tract infection, high blood pressure and bone fractures.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
1. Why am I using ARX-Abiraterone?
ARX-Abiraterone contains the active ingredient abiraterone acetate. ARX-Abiraterone belongs to a class of drugs known as anti-androgens (anti-testosterone). Testosterone, a natural hormone, helps prostate cancer to grow and spread.
ARX-Abiraterone is used to treat prostate cancer that has spread to other parts of the body. It reduces the levels of the sex hormone testosterone.
Ask your doctor if you have any questions about why ARX-Abiraterone has been prescribed for you.
This medicine is available only with a doctor's prescription.
ARX-Abiraterone is not addictive.
2. What should I know before I use ARX-Abiraterone?
Warnings
Do not use ARX-Abiraterone if:
- if you are pregnant or maybe potentially pregnant
ARX-Abiraterone is not for use in women and children. - if you have severe liver failure
ARX-Abiraterone should not be used in patients with severe liver failure. Ask your doctor if you have questions. - you are allergic to abiraterone acetate, or any of the ingredients listed at the end of this leaflet.
Always check the ingredients to make sure you can use this medicine. - if the packaging is torn or shows signs of tampering.
- if the expiry date (month and year) printed on the pack has passed.
- if you have moderate or severe liver disease. Your doctor will decide whether ARX-Abiraterone can be used if you have a mild liver problem.
Check with your doctor if you:
- have or have had any medical conditions, especially the following:
- Problems with your liver
- high blood pressure or
- heart failure or
- low blood potassium.
Taking ARX-Abiraterone can make these conditions worse. Taking prednisolone with ARX-Abiraterone helps to avoid worsening of these conditions. If you have these conditions, or other heart or blood vessel problems, discuss them with your doctor.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and Breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
ARX-Abiraterone must not be taken by women who are pregnant or might be pregnant since ARX-Abiraterone may affect the baby.
If you are having sex with a pregnant woman you need to use a condom. If you are having sex with a woman who can become pregnant you need to use a condom and another effective birth control method.
If you are pregnant or might be pregnant, wear gloves if you need to touch or handle ARX-Abiraterone.
Check with your doctor if you are breastfeeding or intend to start breastfeeding.
ARX-Abiraterone must not be taken by women who breastfeeding since ARX-Abiraterone may affect the baby.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interfere with ARX-Abiraterone and affect how it works.
This may result in greater or lesser effects or even side effects from these medicines/treatments.
Your doctor will prescribe prednisolone to reduce the chance of high blood pressure, heart effects or low blood potassium. You will need to take prednisolone daily while you are taking ARX-Abiraterone. Do not stop taking prednisolone unless your doctor tells you to do this. During a medical emergency the dose of prednisolone may need to be increased. Your doctor will decide if this is necessary.
Your doctor may prescribe other treatments to be continued while you are taking ARX-Abiraterone and prednisolone.
If you have diabetes, your blood sugar may drop if you take ARX-Abiraterone plus prednisolone with some medicines for diabetes such as pioglitazone or repaglinide. Tell your doctor if you notice a drop in your blood sugar.
4. How do I use ARX-Abiraterone?
How much to take
- Always take ARX-Abiraterone exactly as your doctor has told you. You must check with your doctor if you are not sure.
- The usual daily dose of ARX-Abiraterone is four 250 mg tablets or two 500 mg tablets taken as a single dose.
When to take:
- Take as a single dose once daily on an empty stomach. ARX-Abiraterone must be taken at least one hour before or two hours after eating food
- Do not take ARX-Abiraterone with food.
- Taking ARX-Abiraterone with food causes more of this medicine to be absorbed by the body than is needed and this may cause side effects.
- ARX-Abiraterone tablets should be swallowed whole with water. Do not break the tablets.
- ARX-Abiraterone is prescribed with prednisolone. The usual dose of prednisolone is 5 or 10 mg daily taken according to your doctor's instructions.
- You may also be required to take, or continue taking, additional hormone therapy. Follow all the instructions provided by your doctor. Ask your doctor or pharmacist if you have any questions.
If you forget to use ARX-Abiraterone
ARX-Abiraterone should be used regularly at the same time each day.
If you miss a daily dose of ARX-Abiraterone or prednisolone, take your normal dose the following day. If more than one daily dose is missed, talk to your doctor.
If you use too much ARX-Abiraterone
If you think that you have used too much ARX-Abiraterone, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre (by calling 13 11 26), or
- contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using ARX-Abiraterone?
Things you should do
Remind any doctor, dentist or pharmacist you visit that you are using ARX-Abiraterone. If you are undergoing anaesthesia, tell your anaesthetist that you are taking ARX-Abiraterone
Do not stop taking ARX-Abiraterone unless your doctor tells you to do so.
Be sure to keep all your doctor's appointments so your progress can be checked.
Call your doctor straight away if you:
Notice any of these signs of low blood potassium:
- muscle weakness
- muscle twitches
- heart palpitations
If you are about to be started on any new medicines, tell your doctor or pharmacist that you are taking ARX-Abiraterone.
If you have any further questions on the use of this product, ask your doctor.
Driving or using machines
Be careful driving or operating machinery until you know how ARX-Abiraterone affects you.
Looking after your medicine
Follow the instructions on how to take care of your medicine properly. Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills
250 mg tablet: Store below 25°C
500 mg tablet: Store below 30°C
Keep it where young children cannot reach it.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
Cardiovascular Symptoms:
| Call your doctor straight away or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What ARX-Abiraterone contains
| Active ingredient (main ingredient) | abiraterone acetate |
| Other ingredients (inactive ingredients) | lactose monohydrate microcrystalline cellulose croscarmellose sodium sodium lauryl sulfate magnesium stearate hypromellose colloidal silicon dioxide. Opadry II Purple 85F500067 |
| Potential allergens | Lactose |
Do not take this medicine if you are allergic to any of these ingredients.
What ARX-Abiraterone looks like
ARX-Abiraterone 250 mg tablet is white to off white, oval shaped, film coated tablets, debossed with “35” on one side and plain on other side. (AUST R 490407).
ARX-Abiraterone 500 mg tablet is Pinkish brown color, oval shaped, biconvex, film coated tablets, debossed with "46" on one side and plain other side (AUST R 490408).
Who distributes ARX-Abiraterone
Arrotex Pharmaceuticals Pty Ltd
15 – 17 Chapel Street
Cremorne VIC 3121
Australia
www.arrotex.com.au
This leaflet was prepared in August 2025.
Brand Information
| Brand name | ARX-Abiraterone |
| Active ingredient | Abiraterone acetate |
| Schedule | S4 |
MIMS Revision Date: 01 December 2025
1 Name of Medicine
Abiraterone acetate.
2 Qualitative and Quantitative Composition
Each 250 mg tablet contains 250 mg of abiraterone acetate.
Each 500 mg tablet contains 500 mg abiraterone acetate.
Excipient(s) with known effect. Contains sugars as lactose.
The 250 mg tablets contain 198.65 mg of lactose monohydrate.
The 500 mg tablets contain 253.2 mg of lactose monohydrate.
For a full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
ARX-Abiraterone 250 mg tablets are white to off-white, oval shaped film coated tablets, debossed with "35" on one side and plain on the other side.
ARX-Abiraterone 500 mg film-coated tablets are pinkish brown colour, oval-shaped, biconvex film coated tablets, debossed with "46" on one side and plain on the other side.
4 Clinical Particulars
4.1 Therapeutic Indications
ARX-Abiraterone is indicated in combination with prednisolone for the treatment of:
newly diagnosed high-risk metastatic hormone sensitive prostate cancer (mHSPC) in combination with androgen deprivation therapy (ADT); or
patients with metastatic advanced prostate cancer (castration resistant prostate cancer, mCRPC) who are asymptomatic or mildly symptomatic after failure of androgen deprivation therapy (ADT); or
patients with mCRPC who have received prior chemotherapy containing a taxane.
4.2 Dose and Method of Administration
The recommended dosage of ARX-Abiraterone is 1 g (four 250 mg tablets or two 500 mg tablets) as a single daily dose that must not be taken with food. ARX-Abiraterone tablets must be taken as a single dose once daily on an empty stomach. ARX-Abiraterone must be taken at least two hours after eating and food must not be eaten for at least one hour after taking ARX-Abiraterone. The tablets must be swallowed whole with water (see Section 5.2 Pharmacokinetic Properties, Absorption).
Dosage of prednisolone. For hormone sensitive prostate cancer (mHSPC), ARX-Abiraterone is used with 5 mg prednisolone daily (see Section 4.4 Special Warnings and Precautions for Use, Corticosteroid withdrawal and coverage of stress situations).
For metastatic castration-resistant prostate cancer (mCRPC), ARX-Abiraterone is used with 10 mg prednisolone daily (see Section 4.4 Special Warnings and Precautions for Use, Corticosteroid withdrawal and coverage of stress situations).
Recommended monitoring. Serum transaminases and bilirubin should be measured prior to starting treatment with ARX-Abiraterone, every two weeks for the first three months of treatment and monthly thereafter. Blood pressure, serum potassium and fluid retention should be monitored monthly (see Section 4.4 Special Warnings and Precautions for Use).
Patients started on ARX-Abiraterone who were receiving a LHRH agonist should continue to receive a LHRH agonist.
Renal insufficiency. No dosage adjustment is necessary for patients with renal impairment.
Hepatic insufficiency. No dosage adjustment is necessary for patients with pre-existing mild hepatic impairment. There are no data on the clinical safety and efficacy of multiple doses of abiraterone acetate when administered to patients with moderate or severe hepatic impairment (Child-Pugh Class B or C). No dose adjustment can be predicted. ARX-Abiraterone should be used with caution in patients with moderate hepatic impairment, only if the benefit clearly outweighs the possible risk. ARX-Abiraterone should not be used in patients with pre-existing severe hepatic impairment (see Section 4.3 Contraindications; Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties).
For patients who develop hepatotoxicity during treatment with ARX-Abiraterone (alanine aminotransferase (ALT) or aspartate aminotransferase (AST) increases above 5 times the upper limit of normal or bilirubin increases above 3 times the upper limit of normal) treatment should be withheld immediately until liver function tests normalise (see Section 4.4 Special Warnings and Precautions for Use). Re-treatment following return of liver function tests to the patient's baseline may be given at a reduced dose of 500 mg (one 500 mg tablet or two 250 mg tablets) once daily. For patients being retreated, serum transaminases and bilirubin should be monitored at a minimum of every two weeks for three months and monthly thereafter. If hepatotoxicity recurs at the reduced dose of 500 mg daily, discontinue treatment with ARX-Abiraterone. Reduced doses should not be taken with food.
If patients develop severe hepatotoxicity (ALT or AST 20 times the upper limit of normal) anytime while on therapy, ARX-Abiraterone should be discontinued and patients should not be re-treated with ARX-Abiraterone.
4.3 Contraindications
ARX-Abiraterone is contraindicated in women who are or may potentially be pregnant.
ARX-Abiraterone is contraindicated in patients with severe hepatic impairment [Child Pugh Class C] (see Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties).
ARX-Abiraterone plus prednisolone is contraindicated in combination with Xofigo (radium 223 dichloride; see Section 4.4).
4.4 Special Warnings and Precautions for Use
Hypertension, hypokalaemia and fluid retention due to mineralocorticoid excess. ARX-Abiraterone should be used with caution in patients with a history of cardiovascular disease. The safety of abiraterone acetate in patients with left ventricular ejection fraction (LVEF) < 50% or New York Heart Association (NYHA) Class III or IV heart failure (in study 301) or NYHA Class II to IV heart failure (in studies 3011 and 302) was not established. Before treatment with ARX-Abiraterone, hypertension must be controlled, and hypokalaemia must be corrected.
Abiraterone may cause hypertension, hypokalaemia and fluid retention (see Section 4.8 Adverse Effects (Undesirable Effects)) as a consequence of increased mineralocorticoid levels resulting from CYP17 inhibition (see Section 5.1 Pharmacodynamic Properties). Co-administration of a corticosteroid suppresses adrenocorticotropic hormone (ACTH) drive, resulting in a reduction in the incidence and severity of these adverse reactions. Caution is required in treating patients whose underlying medical conditions might be compromised by increases in blood pressure, hypokalaemia or fluid retention, e.g. those with heart failure, recent myocardial infarction or ventricular arrhythmia. In postmarketing experience, QT prolongation and Torsades de Pointes have been observed in patients who develop hypokalaemia or have underlying cardiovascular conditions while taking abiraterone.
Blood pressure, serum potassium and fluid retention should be monitored at least monthly.
Hepatotoxicity. Marked increases in liver enzymes leading to drug discontinuation or dosage modification occurred in controlled clinical studies (see Section 4.8 Adverse Effects (Undesirable Effects)). Very rarely hepatitis fulminant and hepatic failure has been seen. Serum transaminase and bilirubin levels should be measured prior to starting treatment with abiraterone, every two weeks for the first three months of treatment, and monthly thereafter. If clinical symptoms or signs suggestive of hepatotoxicity develop, serum transaminases, should be measured immediately. If at any time the ALT or AST rises above 5 times the upper limit of normal or the bilirubin rises above 3 times the upper limit of normal, treatment with abiraterone should be interrupted immediately and liver function closely monitored.
Re-treatment with abiraterone may only take place after the return of liver function tests to the patient's baseline and at a reduced dose level (see Section 4.2 Dose and Method of Administration).
If patients develop severe hepatotoxicity (ALT or AST 20 times the upper limit of normal) anytime while on therapy, abiraterone should be discontinued and patients should not be re-treated with abiraterone.
Patients with active or symptomatic viral hepatitis were excluded from clinical trials; thus, there are no data to support the use of abiraterone acetate in this population.
There are no data on the clinical safety and efficacy of multiple doses of abiraterone acetate when administered to patients with moderate or severe hepatic impairment (Child-Pugh Class B or C). The use of ARX-Abiraterone should be cautiously assessed in patients with moderate hepatic impairment, in whom the benefit clearly should outweigh the possible risk. ARX-Abiraterone should not be used in patients with severe hepatic impairment (see Section 4.3 Contraindications; Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties).
Corticosteroid withdrawal and coverage of stress situations. Caution is advised and monitoring for adrenocortical insufficiency should occur if patients need to be withdrawn from prednisolone. If abiraterone is continued after corticosteroids are withdrawn, patients should be monitored for symptoms of mineralocorticoid excess.
In patients on prednisolone who are subjected to unusual stress, increased dosage of a corticosteroid may be indicated before, during and after the stressful situation. 17α hydroxylase inhibition by abiraterone decreases glucocorticoid production.
Hyperglycaemia. The use of glucocorticoids could increase hyperglycaemia, therefore blood sugar should be measured frequently in patients with diabetes.
Hypoglycaemia. Isolated cases of hypoglycaemia have been reported when abiraterone plus prednisone*/prednisolone was administered to patients with pre-existing diabetes receiving pioglitazone or repaglinide (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Blood glucose should be monitored in patients with diabetes.
Use with chemotherapy. The safety and efficacy of concomitant use of abiraterone with cytotoxic chemotherapy has not been established.
Use in combination with radium 223 dichloride. In a randomised clinical trial in patients with asymptomatic or mildly symptomatic bone-predominant metastatic castration resistant prostate cancer, at the time of unblinding, the addition of radium 223 dichloride to abiraterone plus *prednisone/prednisolone showed an increase in mortality and an increased rate of fracture. Radium 223 dichloride is not recommended for use in combination with ARX-Abiraterone plus prednisolone. It is recommended that subsequent treatment with radium 223 dichloride is not initiated for at least 5 days after the last administration of ARX-Abiraterone in combination with prednisolone.
Use in the elderly. No data available.
Paediatric use. This medicine is not for use in children.
Effects on laboratory tests. No data available.
* All safety data from studies co-administering abiraterone with prednisone alone or prednisone/prednisolone have been included in the PI to ensure completeness of safety information. This safety information is applicable to coadministration of ARX-Abiraterone with prednisolone.
4.5 Interactions with Other Medicines and Other Forms of Interactions
In vitro studies. In vitro studies with human hepatic microsomes showed that abiraterone is a strong inhibitor of CYP1A2, CYP2D6 and CYP2C8 and a moderate inhibitor of CYP2C9, CYP2C19 and CYP3A4/5. The major metabolites, abiraterone sulphate and N-oxide abiraterone sulphate are also strong inhibitors of CYP2C8 in vitro. In vitro abiraterone, abiraterone sulphate and N-oxide abiraterone sulphate are inhibitors of OATP1B1. The clinical relevance of this inhibition is not clear.
Clinical studies. CYP2D6. In a study to determine the effects of abiraterone acetate (plus prednisone) on a single dose of the CYP2D6 substrate dextromethorphan, the systemic exposure (AUC) of dextromethorphan was increased approximately 200%. The AUC24 for dextrorphan, the active metabolite of dextromethorphan, increased approximately 33%.
Caution is advised when abiraterone is administered with drugs activated by or metabolised by CYP2D6, particularly with drugs that have a narrow therapeutic index. Dose reduction of narrow therapeutic index drugs metabolised by CYP2D6 should be considered the same.
CYP3A4. Abiraterone is a substrate of CYP3A4. In a clinical pharmacokinetic interaction study of 20 healthy subjects pre-treated with a strong CYP3A4 inducer (rifampicin, 600 mg daily for 6 days) followed by a single dose of abiraterone acetate 1000 mg, the mean plasma Cmax and AUC∞ of abiraterone were decreased by 55%.
Strong inducers of CYP3A4 (e.g. phenytoin, carbamazepine, rifampicin, rifabutin, rifapentine, phenobarbital) during treatment with ARX-Abiraterone is to be avoided, or used with careful evaluation of clinical efficacy, if there is no therapeutic alternative.
In a separate clinical pharmacokinetic interaction study of 19 healthy subjects, co-administration of ketoconazole, a strong inhibitor of CYP3A4 (ketoconazole 400 mg for 6 days), had no clinically meaningful effect on the pharmacokinetics of abiraterone.
CYP2C8. In a CYP2C8 drug-drug interaction trial in healthy subjects, the AUC of pioglitazone was increased by 46% and the AUCs for M-III and M-IV, the active metabolites of pioglitazone, each decreased by 10%, when pioglitazone was given together with a single dose of 1000 mg abiraterone acetate.
Patients should be monitored for signs of toxicity related to a CYP2C8 substrate with a narrow therapeutic index if used concomitantly with ARX-Abiraterone. Examples of medicinal products metabolized by CYP2C8 include pioglitazone and repaglinide (see Section 4.4 Special Warnings and Precautions for Use, Hypoglycaemia).
CYP1A2. In a clinical study to determine the effects of abiraterone acetate (plus prednisone) on a single dose of the CYP1A2 substrate theophylline, no increase in systemic exposure of theophylline was observed.
Use with spironolactone. Spironolactone binds to the androgen receptor and may increase prostate specific antigen (PSA) levels. Use with ARX-Abiraterone is not recommended (see Section 5.1 Pharmacodynamic Properties, Pharmacodynamic effects).
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. In fertility studies in both male and female rats (4-and 3-weeks), abiraterone acetate reduced fertility, which was completely reversible in 4 to 16 weeks after abiraterone acetate was stopped.
In studies in mice (4 weeks), rats (4 up to 26-weeks) and monkeys (up to 39-weeks), decreases in testosterone levels, atrophy, aspermia/hypospermia, and/or hyperplasia in the reproductive system were observed at > 125 mg/kg/day in mice, ≥ 30 mg/kg/day in rats and ≥ 250 mg/kg/day in monkeys and were consistent with the antiandrogenic pharmacological activity of abiraterone. These effects were observed at exposure levels similar to or lower than the human clinical exposure, based on abiraterone AUC.
Use in pregnancy. (Category D)
ARX-Abiraterone is contraindicated in women who are or may potentially be pregnant (see Section 4.3 Contraindications).
There are no human data on the use of abiraterone in pregnancy and abiraterone is not for use in women of child-bearing potential. Maternal use of a CYP17 inhibitor is expected to produce changes in hormone levels that could affect development of the fetus.
In an embryofetal developmental study in the rat, abiraterone acetate at ≥ 10 mg/kg/day affected pregnancy including reduced fetal weight and survival, delayed ossification, and increases in late resorptions and post implantation loss with a subsequent reduction in live fetuses. Effects on the external genitalia (decreased fetal ano-genital distance) were observed though abiraterone acetate was not teratogenic.
In these fertility and developmental toxicity studies performed in the rat, all effects were related to the pharmacological activity of abiraterone.
It is not known if abiraterone or its metabolites are present in semen. A condom is required if the patient is engaged in sexual activity with a pregnant woman. If the patient is engaged in sex with a woman of child-bearing potential, a condom is required along with another effective contraceptive method.
To avoid inadvertent exposure, women who are pregnant or women who may be pregnant should not handle ARX-Abiraterone without protection, e.g. gloves.
Use in lactation. ARX-Abiraterone is not for use in women.
It is not known if either abiraterone or its metabolites are excreted in human breast milk.
4.7 Effects on Ability to Drive and Use Machines
No studies on the effects of abiraterone on the ability to drive or use machines have been performed. It is not anticipated that abiraterone will affect the ability to drive and use machines.
4.8 Adverse Effects (Undesirable Effects)
Adverse drug reactions from clinical trials. In an analysis of adverse reactions of composite Phase 3 studies with abiraterone, adverse reactions that were observed in ≥ 10% of patients were peripheral oedema, hypokalaemia, hypertension, urinary tract infection, and alanine aminotransferase increased, and/or aspartate aminotransferase increased.
Abiraterone may cause hypertension, hypokalaemia and fluid retention as a pharmacodynamic consequence of its mechanism of action. In Phase 3 studies anticipated mineralocorticoid effects were seen more commonly in patients treated with abiraterone versus patients treated with placebo; hypokalaemia 18% versus 8%, hypertension 22% versus 16% and fluid retention (peripheral oedema) 23% versus 17%, respectively. Grades 3/4 hypokalaemia were observed in 6% versus 1%, grades 3/4 hypertension were observed in 7% versus 5%, and grades 3/4 fluid retention oedema were observed in 1% versus 1% of patients treated with abiraterone versus patients treated with placebo, respectively. Mineralocorticoid effects generally were able to be successfully managed medically. Concomitant use of a corticosteroid reduces the incidence and severity of these adverse drug reactions (see Section 4.4 Special Warnings and Precautions for Use).
In a Phase 3 study of patients with newly diagnosed high-risk mHNPC or mHSPC (Study 3011) who were receiving and remained on ADT (a luteinising hormone-releasing hormone [LHRH] agonist or orchiectomy), and abiraterone acetate was administered at a dose of 1000 mg daily in combination with low dose prednisone (5 mg daily) and ADT in the active treatment arm; ADT and placebo were given to control patients. The median duration of treatment with abiraterone was 24 months.
Adverse reactions that occurred at a rate of ≥ 1% (all grades) are shown in Table 1.
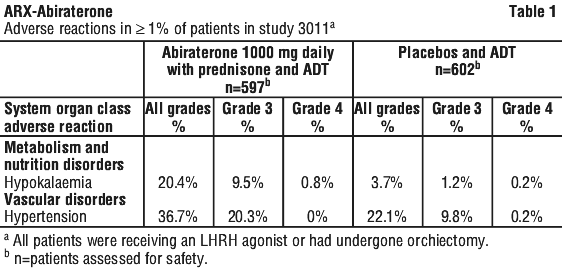
Adverse drug reactions that occurred at a rate of ≥ 1% (all grades) are shown in Table 2.
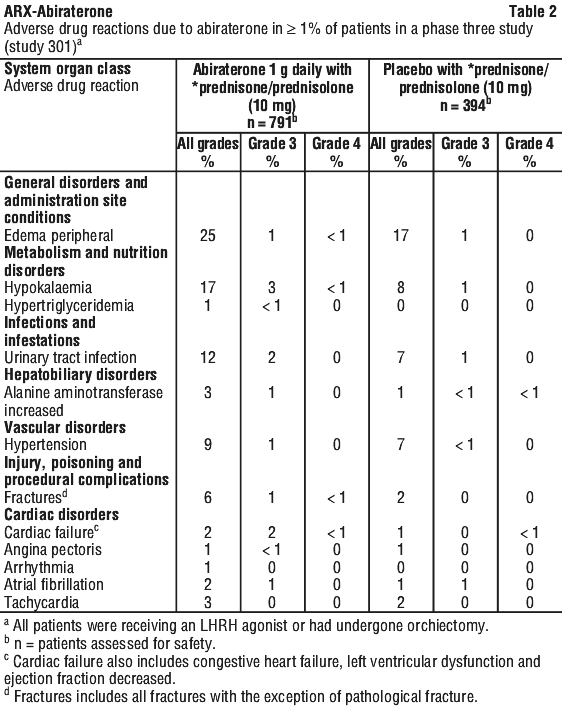
Adverse drug reactions that occurred at a rate of ≥ 1% (all grades) are shown in Table 3.
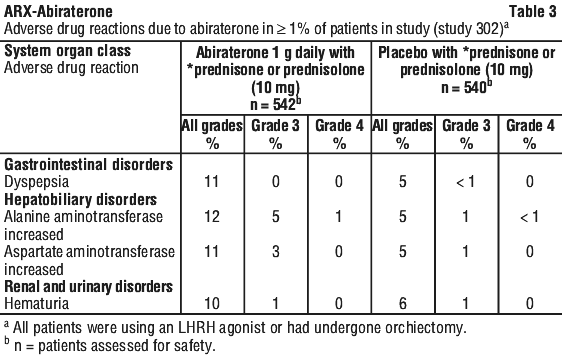
The adverse drug reaction, adrenal insufficiency, occurred in the Phase 3 clinical studies at a rate 0.3% in patients taking abiraterone and at a rate of 0.1% inpatients taking placebo.
In the Phase 3 studies, 70% of patients were 65 years and over, and 27% were 75 years and over for patients taking abiraterone. Adverse effects were more common in patients ≥ 75 years old in both the abiraterone and placebo groups.
Cardiovascular effects. The Three Phase 3 studies excluded patients with uncontrolled hypertension, clinically significant heart disease as evidenced by myocardial infarction, arterial thrombotic events in the past 6 months, severe or unstable angina, or NYHA Class III or IV heart failure (study 301) or Class II to IV heart failure (studies 3011 and 302) or cardiac ejection fraction measurement of < 50%. All patients enrolled (both active and placebo-treated patients) were concomitantly treated with androgen deprivation therapy, predominately with the use of LHRH agonists, which has been associated with diabetes, myocardial infarction, cerebrovascular accident and sudden cardiac death. The incidence of cardiovascular adverse reactions in the Phase 3 studies in patients taking abiraterone versus patients taking placebo were as follows: atrial fibrillation 2.6% vs. 2.0%, tachycardia 1.9% vs. 1.0%, angina pectoris 1.7% vs. 0.8%, cardiac failure 0.7% vs. 0.2% and arrhythmia 0.7% vs. 0.5%.
Hepatotoxicity. Drug-associated hepatotoxicity with elevated ALT, AST and total bilirubin has been reported in patients treated with abiraterone. Across Phase 3 clinical studies, hepatotoxicity grades 3 and 4 (e.g. ALT or AST increases of > 5 X ULN or bilirubin increases > 1.5 X ULN) were reported in approximately 4% of patients who received abiraterone, typically during the first 3 months after starting treatment. In Study 3011, grade 3 or 4 hepatotoxicity was observed in 8.4% of patients treated with abiraterone acetate. Ten patients who received abiraterone acetate were discontinued because of hepatotoxicity; two had Grade 2 hepatotoxicity, six had Grade 3 hepatotoxicity, and two had Grade 4 hepatotoxicity. No patient died of hepatotoxicity in Study 3011. In the 301 clinical study, patients whose baseline ALT or AST were elevated were more likely to experience liver function test elevations than those beginning with normal values. When elevations of either ALT or AST > 5 X ULN, or elevations in bilirubin > 3 X ULN were observed, abiraterone was withheld or discontinued.
Hepatic metastases and baseline elevations in alkaline phosphatase associated with prostate cancer were present in a few of these patients. In two instances marked increases in liver function tests occurred (see Section 4.4 Special Warnings and Precautions for Use). These two patients with normal baseline hepatic function, experienced ALT or AST elevations 15 to 40 X ULN and bilirubin elevations 2 to 6 X ULN. Upon discontinuation of abiraterone, both patients had normalisation of their liver function tests and one patient was re-treated with abiraterone without recurrence of the elevations. In study 302, grade 3 or 4 ALT or AST elevations were observed in 35 (6.5%) patients treated with abiraterone. Aminotransferase elevations resolved in all but 3 patients (2 with new multiple liver metastases and 1 with AST elevation approximately 3 weeks after the last dose of abiraterone). In Phase 3 clinical studies, treatment discontinuations due to ALT and AST increases or abnormal hepatic function were reported in 1.1% of patients treated with abiraterone and 0.6% of patients treated with placebo. No deaths were reported due to hepatotoxicity events.
In clinical trials, the risk for hepatotoxicity was mitigated by exclusion of patients with baseline hepatitis or significant abnormalities of liver function tests. In the 3011 trial, patients with baseline ALT and AST > 2.5 X ULN, bilirubin > 1.5 X ULN or those with active or symptomatic viral hepatitis or chronic liver disease; ascites or bleeding disorders secondary to hepatic dysfunction were excluded. In the 301 trial, patients with baseline ALT and AST ≥ 2.5X ULN in the absence of liver metastases and > 5 X ULN in the presence of liver metastases were excluded. In the 302 trial patients with liver metastases were not eligible and patients with baseline ALT and AST ≥ 2.5 X ULN were excluded. Abnormal liver function tests developing in patients participating in clinical trials were vigorously managed by requiring treatment interruption and permitting re-treatment only after return of liver function tests to the patient's baseline (see Section 4.2 Dose and Method of Administration). Patients with elevations of ALT or AST > 20X ULN were not re-treated. The safety of re-treatment in such patients is unknown. The mechanism for hepatotoxicity associated with abiraterone is not understood.
Post-marketing data. Adverse drug reactions identified during the post-marketing experience based on spontaneous reports with abiraterone acetate are described below. The frequencies are provided according to the following convention:
Very common ≥ 1/10; common ≥ 1/100 and < 1/10; uncommon ≥ 1/1,000 and < 1/100; rare ≥ 1/10,000 and < 1/1,000; very rare < 1/10,000; isolated reports: frequency unknown.
System organ class. Respiratory, thoracic and mediastinal disorders. Rare: Allergic alveolitis.
System organ class. Musculoskeletal and connective tissue disorders. Uncommon: Rhabdomyolysis, myopathy.
System organ class. Gastrointestinal disorders. Very common: Diarrhoea.
System organ class. Hepatobiliary disorders. Very rare: Hepatitis fulminant, hepatic failure.
System organ class. Cardiac disorders. Very rare: QT prolongation and Torsades de Pointes (observed in patients who developed hypokalaemia or had underlying cardiovascular conditions).
System organ class. Immune system disorders - hypersensitivity. Very rare: Anaphylactic reaction (severe allergic reactions that include, but are not limited to difficulty swallowing or breathing, swollen face, lips, tongue or throat, or an itchy rash (urticaria)).
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
* All safety data from studies co-administering abiraterone with prednisone alone or prednisone/prednisolone have been included in the PI to ensure completeness of safety information. This safety information is applicable to coadministration of ARX-Abiraterone with prednisolone.
4.9 Overdose
There have been no reports of overdose of abiraterone acetate during clinical studies.
There is no specific antidote. In the event of an overdose, administration of abiraterone acetate should be stopped and general supportive measures undertaken, including monitoring for arrhythmias. Liver function also should be assessed.
Contact the Poisons Information Centre (telephone 131126) for advice on management of overdose.
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action. Abiraterone acetate is converted in vivo to abiraterone, an androgen biosynthesis inhibitor. Specifically, abiraterone selectively inhibits the enzyme 17α hydroxylase/C17,20-lyase (CYP17). This enzyme is expressed in and is required for androgen biosynthesis in testicular, adrenal and in prostatic tumour tissues. It catalyses the conversion of pregnenolone and progesterone into testosterone precursors, DHEA and androstenedione, respectively, by 17α hydroxylation and cleavage of the C17,20 bond. CYP17 inhibition also results in increased mineralocorticoid production by the adrenals (see Section 4.4 Special Warnings and Precautions for Use).
Androgen-sensitive prostatic carcinoma responds to treatment that decreases androgen levels. Androgen deprivation therapies, such as treatment with luteinising hormone-releasing hormone (LHRH) agonists or orchiectomy, decrease androgen production in the testes but do not affect androgen production by the adrenals or in the tumour. Treatment with abiraterone decreases serum testosterone to undetectable levels (using commercial assays) when given with LHRH agonists (or orchiectomy).
Pharmacodynamic effects. Abiraterone decreases serum testosterone and other androgens to levels lower than those achieved by the use of LHRH agonists alone or by orchiectomy. Prostate specific antigen (PSA) serves as a biomarker in patients with prostate cancer. In a Phase 3 clinical study of patients who failed prior chemotherapy with taxanes, 29% of patients treated with abiraterone, versus 6% of patients treated with placebo, had at least a 50% decline from baseline in PSA levels.
Use of spironolactone. Patients in pivotal clinical trials with abiraterone acetate were not allowed to use spironolactone as spironolactone binds to the androgen receptor and may increase PSA levels.
Effects on the QT interval. In a cardiovascular safety study in patients with metastatic advanced prostate cancer there were no significant effects of abiraterone acetate on the cardiac QT/QTc interval.
Clinical trials. The efficacy of abiraterone was established in three randomised placebo controlled multicentre Phase 3 clinical studies (studies 3011, 301 and 302) of patients with hormone naïve metastatic prostate cancer and metastatic castration resistant prostate cancer.
Study 3011 enrolled patients who were newly diagnosed (within 3 months of randomisation) mHNPC who had high-risk prognostic factors. High-risk prognosis was defined as having at least 2 of the following 3 risk factors: (1) Gleason score of ≥ 8; (2) presence of 3 or more lesions on bone scan; (3) presence of measurable visceral (excluding lymph node disease) metastasis. In the active arm, abiraterone acetate was administered at a dose of 1 g daily in combination with low dose prednisone 5 mg once daily in addition to ADT (LHRH agonist or orchiectomy), which was the standard of care treatment. Patients in the control arm received ADT and placebos for abiraterone acetate.
Study 302 enrolled patients who were asymptomatic or mildly symptomatic and had not received prior chemotherapy, whereas study 301 enrolled patients who received prior chemotherapy containing a taxane. In both studies patients were using a LHRH agonist or were previously treated with orchiectomy. In the active treatment arms, abiraterone was administered at a dose of 1 g daily in combination with low dose *prednisone/prednisolone 5 mg twice daily. Control patients received placebo and low dose *prednisone/prednisolone 5 mg twice daily.
Because changes in PSA serum concentration do not always predict clinical benefit, in all studies patients were maintained on abiraterone until specific discontinuation criteria were met for each study below.
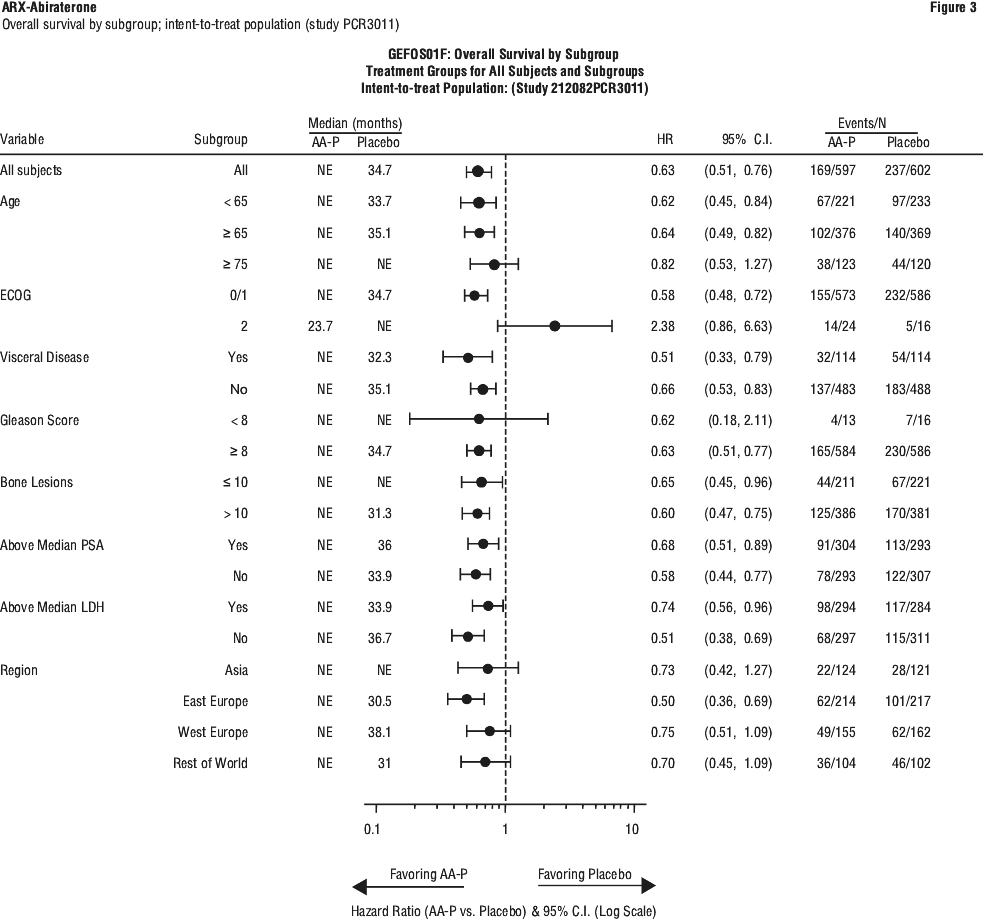
Study 3011 (patients with newly diagnosed high-risk metastatic hormone naïve prostate cancer (mHNPC) or hormone sensitive prostate cancer (mHSPC). In Study 3011, (n=1199) the median age of enrolled patients was 67 years. The ECOG performance status was 0 or 1 for 97% of patients. Patients with uncontrolled hypertension, significant heart disease, or NYHA Class II or worse heart failure were excluded. Co-primary efficacy endpoints were overall survival (OS) and radiographic progression-free survival (rPFS). The median baseline pain score, as measured by the Brief Pain Inventory Short Form (BPI-SF) was 2.0 in both the treatment and placebo groups. In addition to the co primary endpoint measures, benefit was also assessed using time to skeletal-related event (SRE), time to subsequent therapy for prostate cancer, time to initiation of chemotherapy, time to pain progression and time to PSA progression.
In the 3011 study, treatment continued until disease progression, withdrawal of consent, the occurrence of unacceptable toxicity, or death.
Radiographic progression-free survival was defined as the time from randomisation to the occurrence of radiographic progression or death from any cause. Radiographic progression included progression by bone scan (according to modified PCWG2) or progression of soft tissue lesions by CT or MRI (according to RECIST 1.1).
At the planned rPFS analysis there were 593 events; 239 (40.0%) of patients treated with abiraterone acetate and 354 (58.8%) of patients treated with placebo had radiographic evidence of progression or had died. A significant difference in rPFS between treatment groups was observed (see Table 4 and Figure 1).
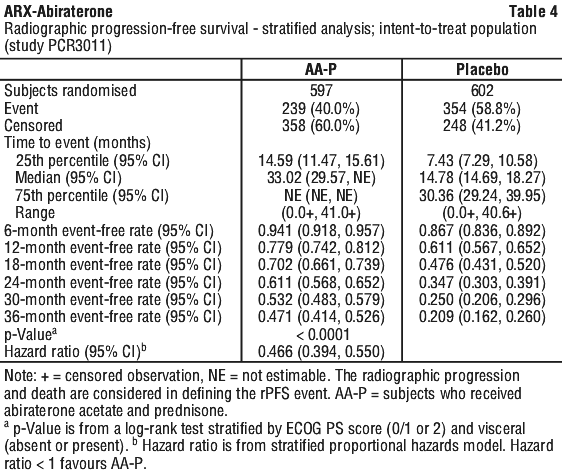
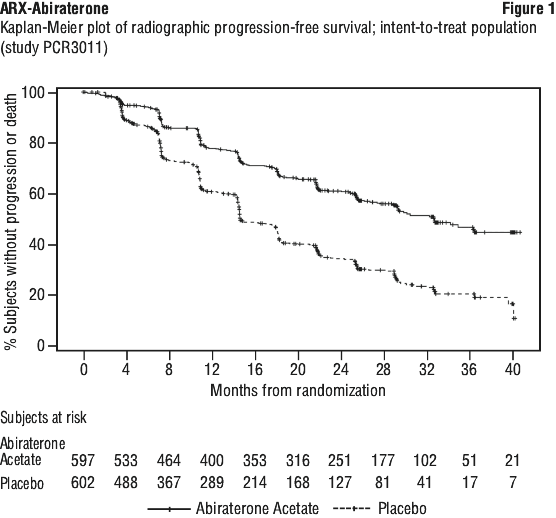
Time to skeletal-related event (SRE). There was a 30% reduction in the risk of skeletal-related events (HR = 0.703; 95% CI: [0.539, 0.916] p < 0.0086). The median time to SRE has not been reached for the abiraterone acetate or placebo study arm.
Time to PSA progression based on PCWG2 criteria. The median time to PSA progression was 33.2 months for patients receiving abiraterone acetate and 7.4 months for patients receiving placebo (HR = 0.299; 95% CI: [0.255, 0.352], p < 0.0001).
Time to subsequent therapy. The median time to subsequent therapy at the time of interim analysis was not reached for patients receiving abiraterone acetate and was 21.6 months for patients receiving placebo (HR = 0.415; 95% CI: [0.346, 0.497], p < 0.0001).
Time to initiation of chemotherapy. The median time to initiation of chemotherapy was not reached for patients receiving abiraterone acetate and was 38.9 months for patients receiving placebo (HR = 0.443; 95% CI: [0.349, 0.561], p < 0.0001).
Time to pain progression. The median time to pain progression was not reached for patients receiving abiraterone acetate and was 16.6 months for patients receiving placebo (HR = 0.695; 95% CI: [0.583, 0.829], p = < 0.0001).
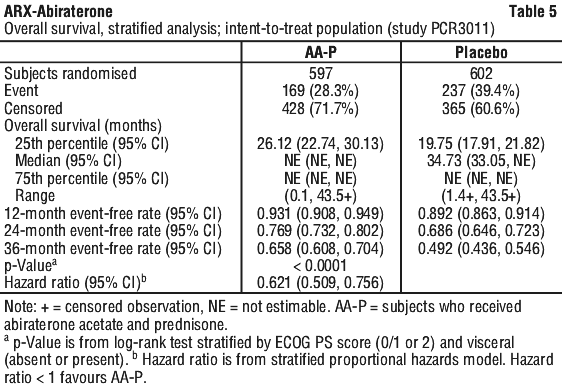
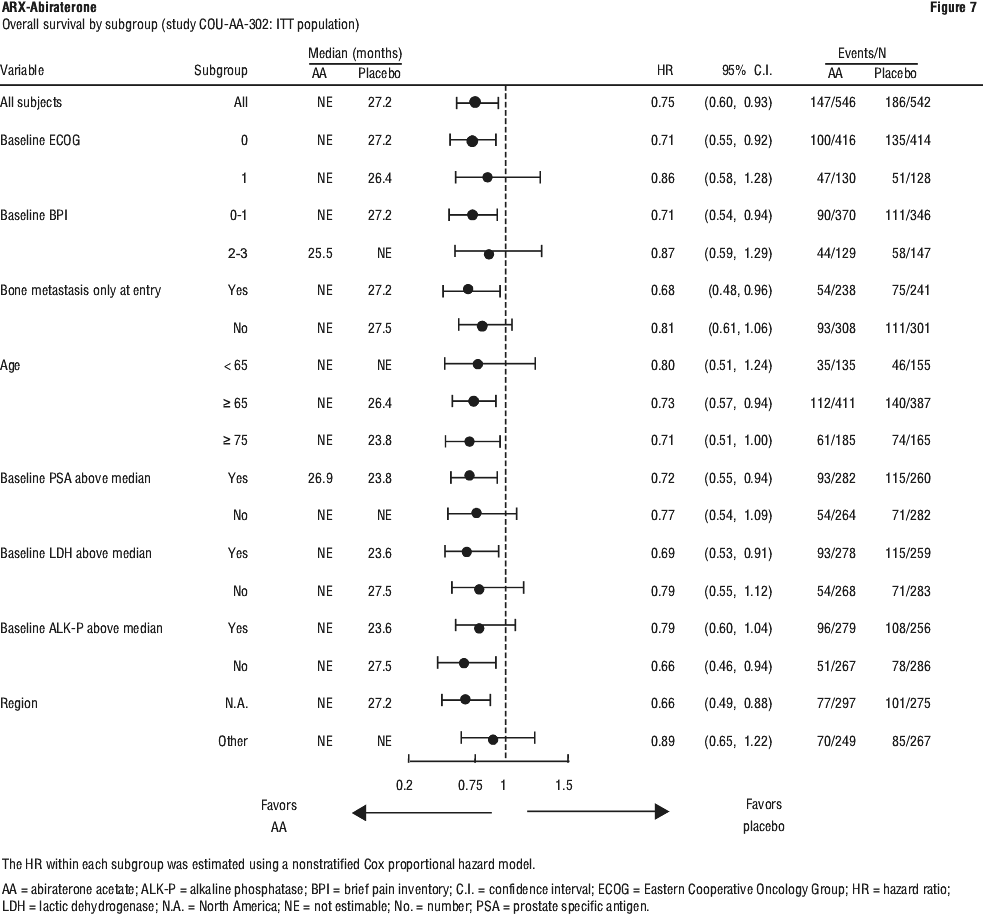
The majority of exploratory endpoints favoured treatment with abiraterone acetate (AA-P) and prednisone over Placebo. A statistically significant improvement in prostate cancer-specific OS was observed for AA-P treatment compared with Placebo (HR=0.547, p < 0.0001). A confirmed PSA response was observed in 91.0% of subjects in the AA-P group and 66.8% of subjects in the Placebo group (relative risk=1.362; p < 0.0001). The overall response rate (complete plus partial response) in subjects with measurable disease at baseline was significantly higher in the AA-P group compared with those in the Placebo group (p=0.0002).
The time to degradation analyses of patient reported outcome (PRO) measures consistently demonstrated that treatment with AA-P delayed degradation and progression of pain, functional status, fatigue and health-related quality of life. Based on the change from baseline using repeated measures mixed-effect model statistically significant differences were observed between AA-P and Placebo as early as Cycle 2 and maintained throughout the study.
Study 302 (asymptomatic or mildly symptomatic patients who did not receive prior chemotherapy). In study 302, (n=1088) the median age of enrolled patients was 71 years for patients treated with abiraterone plus prednisone or prednisolone and 70 years for patients treated with placebo plus prednisone or prednisolone. The ECOG performance status was 0 for 76% of patients, and 1 for 24% of patients in both arms. Co-primary efficacy endpoints were overall survival and radiographic progression-free survival (rPFS). In addition to the co-primary endpoint measures, benefit was also assessed using time to opiate use for cancer pain, time to initiation of cytotoxic chemotherapy, time to deterioration in ECOG performance score by ≥ 1 point and time to PSA progression based on Prostate Cancer Working Group-2 (PCWG2) criteria.
In study 302, treatments were discontinued at the time of unequivocal clinical progression. Treatments could also be discontinued at the time of confirmed radiographic progression at the discretion of the investigator. Patients should not be discontinued based on PSA progression alone and should remain on treatment until fully confirmed clinical progression utilising multiple assessment criteria.
Radiographic progression free survival was assessed with the use of sequential imaging studies as defined by PCWG2 criteria (for bone lesions) and modified Response Evaluation Criteria in Solid Tumours (RECIST) criteria (for soft tissue lesions). PCWG2 criteria require a confirmatory bone scan to document progression. Analysis of rPFS utilised centrally reviewed radiographic assessment of progression.
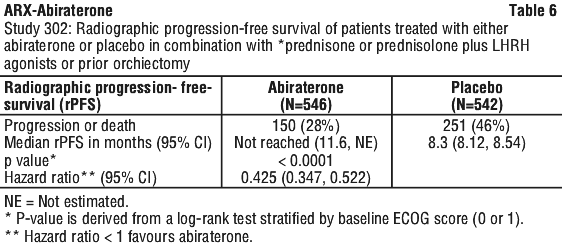
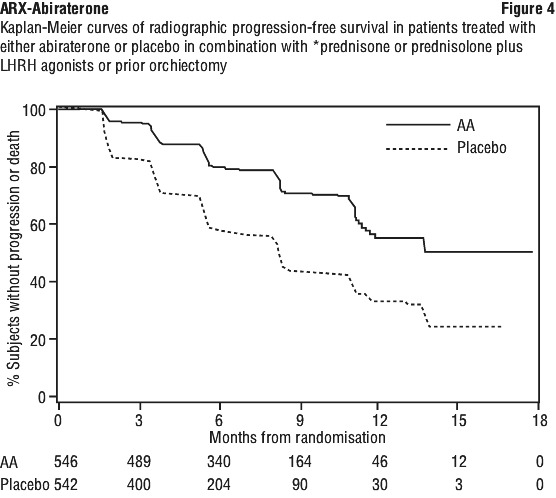
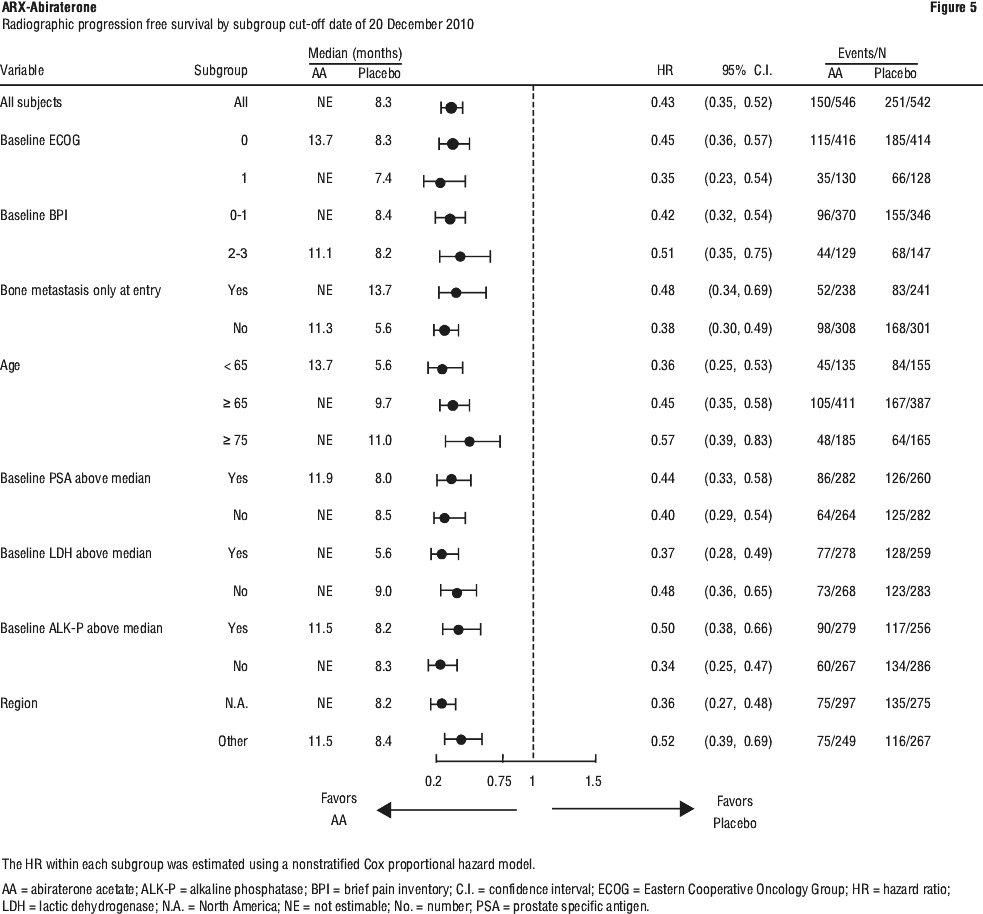
At the planned rPFS analysis there were 401 radiographic progression events; 150 (28%) of patients treated with abiraterone and 251 (46%) of patients treated with placebo had radiographic evidence of progression or had died. A significant difference in rPFS between treatment groups was observed (see Table 6 and Figure 4).
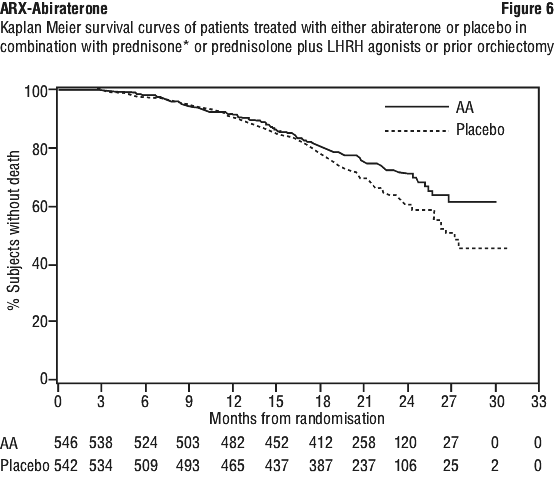
Time to PSA progression based on PCWG2 criteria. Median time to PSA progression was 11.1 months for patients receiving abiraterone and 5.6 months for patients receiving placebo (HR=0.488; 95%CI: [0.420, 0.568], p < 0.0001). Time to PSA progression was approximately doubled with abiraterone treatment. The proportion of subjects with a confirmed PSA response was greater in the abiraterone group than in the placebo group (62% versus 24%; p < 0.0001).
Time to opiate use for cancer pain. The median time to opiate use for prostate cancer pain was not reached for patients receiving abiraterone and was 23.7 months for patients receiving placebo (HR=0.686; 95%CI: [0.566, 0.833], p=0.0001).
Time to initiation of cytotoxic chemotherapy. The median time to initiation of cytotoxic chemotherapy was 25.2 months for patients receiving abiraterone and 16.8 months for patients receiving placebo (HR=0.580; 95% CI: [0.487, 0.691], p < 0.0001).
Time to deterioration in ECOG performance score by ≥ 1 point. The median time to deterioration in ECOG performance score by ≥ 1 point was 12.3 months for patients receiving abiraterone and 10.9 months for patients receiving placebo (HR=0.821; 95% CI: [0.714, 0.943], p=0.0053).
The following study endpoints demonstrated a statistically significant advantage in favour of abiraterone treatment.
Objective response. Objective response was defined as the proportion of subjects with measurable disease achieving a complete or partial response according to RECIST criteria (baseline lymph node size was required to be ≥ 2 cm to be considered a target lesion). The proportion of subjects with measurable disease at baseline who had an objective response was 36% in the abiraterone group and 16% in the placebo group (p < 0.0001).
Pain. Treatment with abiraterone significantly reduced the risk of average pain intensity progression by 18% compared with placebo (p=0.0490). The median time to progression was 26.7 months in the abiraterone group and 18.4 months in the placebo group.
Time to degradation in the FACT-P (total score). Treatment with abiraterone decreased the risk of FACT-P (Total Score) degradation by 22% compared with placebo (p=0.0028). The median time to degradation in FACT-P (total score) was 12.7 months in the abiraterone group and 8.3 months in the placebo group.
Study 301 (patients who had received prior chemotherapy. Eleven percent of patients enrolled in study 301 had an ECOG performance score of 2; 70% had radiographic evidence of disease progression with or without PSA progression; 70% had received one prior cytotoxic chemotherapy and 30% received two. Liver metastasis was present in 11% of patients treated with abiraterone.
It was recommended that patients be maintained on their study drugs until there was PSA progression (confirmed 25% increase over the patient's baseline/nadir) together with protocol-defined radiographic progression and symptomatic or clinical progression. The primary efficacy endpoint was overall survival.
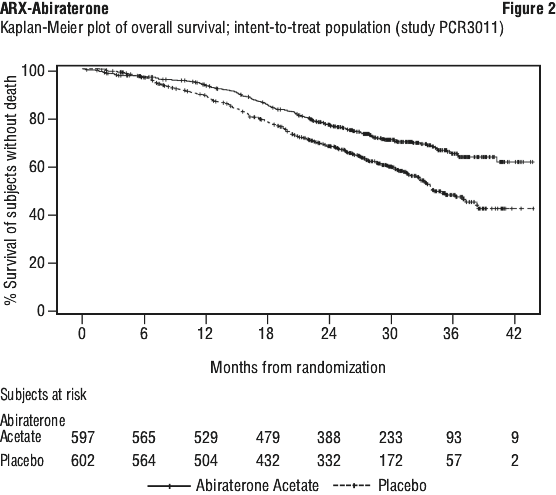
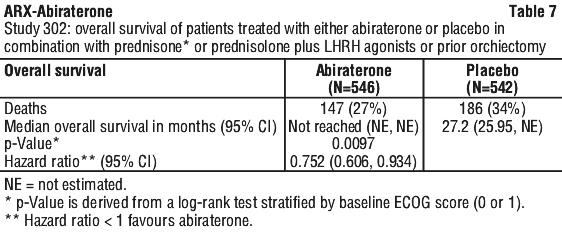
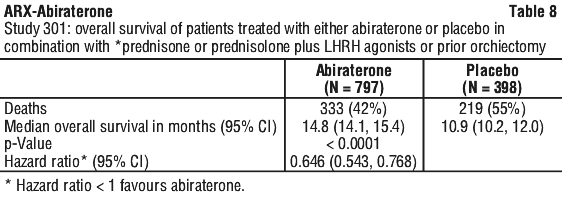
In a planned analysis conducted after 552 deaths were observed, 42% (333 of 797) of patients treated with abiraterone compared with 55% (219 of 398) of patients treated with placebo had died. A statistically significant improvement in median overall survival was seen in patients treated with abiraterone (see Table 8).
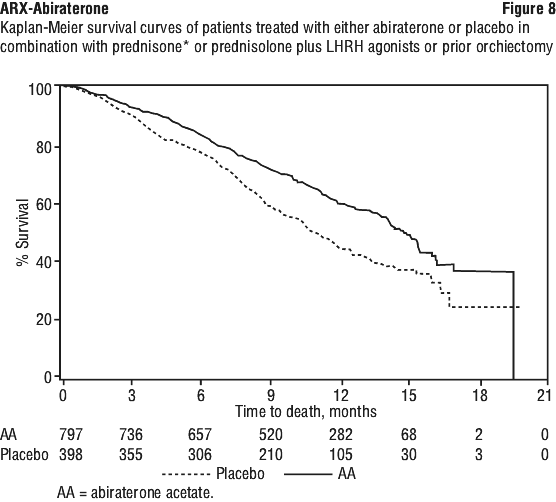
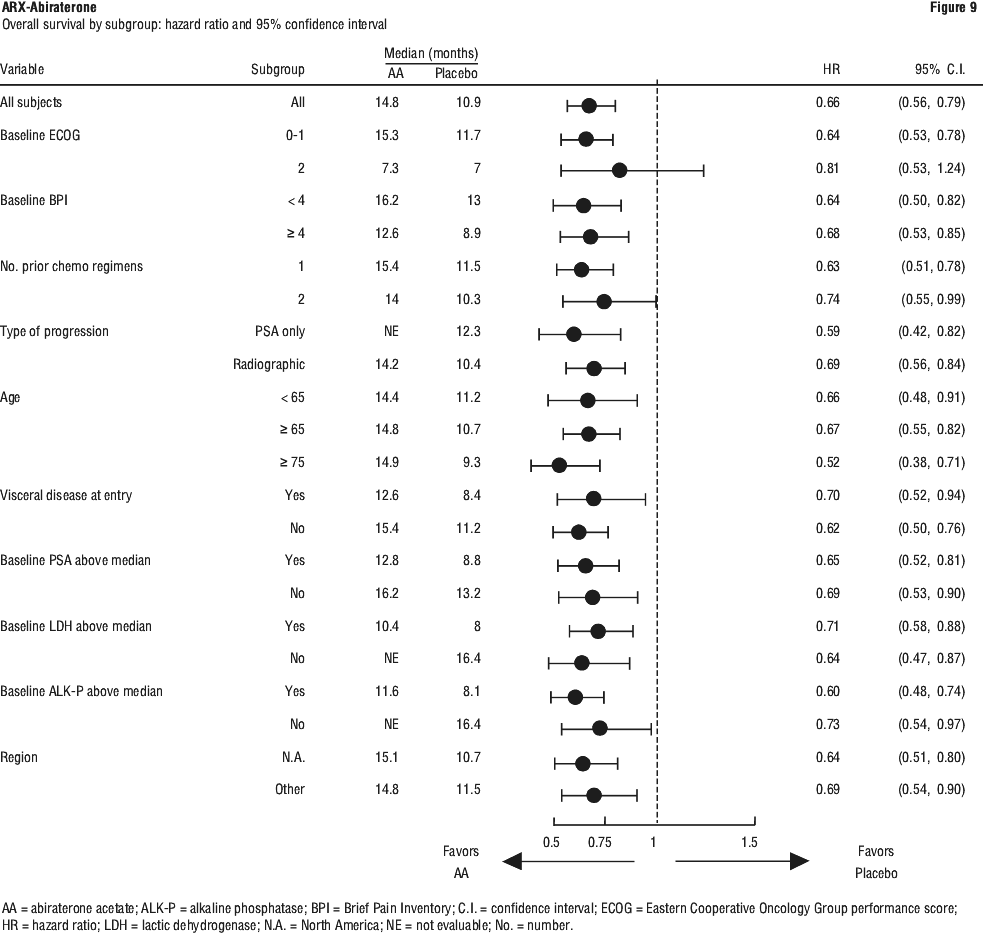
Patients receiving abiraterone demonstrated a significantly higher total PSA response rate (defined as a ≥ 50% reduction from baseline), compared with patients receiving placebo: 29% versus 6%, p < 0.0001.
The median time to PSA progression was 10.2 months for patients treated with abiraterone and 6.6 months for patients treated with placebo (HR= 0.580; 95% CI: [0.462, 0.728], p < 0.0001).
The median radiographic progression free survival was 5.6 months for patients treated with abiraterone and 3.6 months for patients who received placebo (HR= 0.673; 95% CI: [0.585, 0.776], p < 0.0001).
Pain. The proportion of patients with pain palliation was statistically significantly higher in the abiraterone group than in the placebo group (44% versus 27%, p=0.0002).
A lower proportion of patients treated with abiraterone had pain progression compared to patients taking placebo at 6 (22% vs. 28%), 12 (30% vs. 38%) and 18 months (35% vs. 46%). The time to pain progression at the 25th percentile was 7.4 months in the abiraterone group, versus 4.7 months in the placebo group.
Skeletal-related events. A lower proportion of patients in the abiraterone group had skeletal-related events compared with the placebo group at 6 months (18% vs. 28%), 12 months (30% vs 40%), and 18 months (35% vs. 40%). The time to first skeletal-related event at the 25th percentile in the abiraterone group was twice that of the control group at 9.9 months vs 4.9 months.
5.2 Pharmacokinetic Properties
Following administration of abiraterone acetate, the pharmacokinetics of abiraterone has been studied in healthy subjects, patients with metastatic advanced prostate cancer and subjects without cancer with hepatic or renal impairment. Abiraterone acetate is rapidly converted in vivo to abiraterone (see Section 5.1 Pharmacodynamic Properties).
Absorption. Following oral administration of abiraterone acetate in the fasting state, the median time to reach maximum plasma abiraterone concentration is approximately 2 hours.
Effect of food on absorption. Administration of abiraterone acetate with food, compared with administration in a fasted state, results in up to a 17-fold increase in mean systemic exposure of abiraterone depending on the fat content of the meal. Given the normal variation in the content and composition of meals, taking ARX-Abiraterone with meals has the potential to result in highly variable exposures. therefore, ARX-Abiraterone must not be taken with food. ARX-Abiraterone must be taken as a single dose once daily on an empty stomach. ARX-Abiraterone must be taken at least two hours after eating and food must not be eaten for at least one hour after taking ARX-Abiraterone. The tablets must be swallowed whole with water (see Section 4.2 Dose and Method of Administration).
Distribution. The plasma protein binding of 14C abiraterone in human plasma is 99.8%. The apparent volume of distribution is approximately 5630 L, suggesting that abiraterone extensively distributes to peripheral tissues.
Metabolism. Following oral administration of 14C-abiraterone acetate as capsules, abiraterone acetate is hydrolysed to abiraterone, which then undergoes metabolism including sulphation, hydroxylation and oxidation primarily in the liver. The majority of circulating radioactivity (approximately 92%) is found in the form of metabolites of abiraterone. Of 15 detectable metabolites, 2 main metabolites, abiraterone sulphate and N-oxide abiraterone sulphate, each represent approximately 43% of total radioactivity.
The major enzymes involved in the metabolism of abiraterone are CYP3A4 for phase I (oxidative) metabolites, the sulfotransferase (SULT) isozyme SULT2A1, and UDP-glucuronosyl transferase (UGT) UGT1A4. No studies have been conducted to determine if drugs that induce or inhibit these enzymes affect the metabolism of abiraterone.
Excretion. The mean half-life of abiraterone in plasma is approximately 15 hours based on data from healthy subjects. Following oral administration of 14C abiraterone acetate, approximately 88% of the radioactive dose is recovered in faeces and approximately 5% in urine. The major compounds present in faeces are unchanged abiraterone acetate and abiraterone (approximately 55% and 22 % of the administered dose, respectively).
Additional information on special populations. Hepatic impairment. The pharmacokinetics of abiraterone was examined in subjects with pre-existing mild or moderate hepatic impairment (Child-Pugh class A and B, respectively) and in healthy control subjects. Systemic exposure to abiraterone after a single oral 1 g dose increased by approximately 11% and 260% in subjects with mild and moderate pre-existing hepatic impairment, respectively. The mean half-life of abiraterone is prolonged to approximately 18 hours in subjects with mild hepatic impairment and to approximately 19 hours in subjects with moderate hepatic impairment. No dosage adjustment is necessary for patients with pre-existing mild hepatic impairment. There are no data on the clinical safety and efficacy of multiple doses of abiraterone acetate when administered to patients with moderate or severe hepatic impairment (Child-Pugh Class B or C). No dose adjustment can be predicted. ARX-Abiraterone should be used with caution in patients with moderate hepatic impairment, only if the benefit clearly outweighs the possible risk. ARX-Abiraterone should not be used in patients with pre-existing severe hepatic impairment (see Section 4.3 Contraindications; Section 4.4 Special Warnings and Precautions for Use; Section 4.2 Dose and Method of Administration).
For patients who develop hepatotoxicity during treatment with abiraterone suspension of treatment and dosage adjustment may be required (see Section 4.4 Special Warnings and Precautions for Use; Section 4.2 Dose and Method of Administration).
Renal impairment. The pharmacokinetics of abiraterone was compared in patients with end-stage renal disease on a stable haemodialysis schedule versus matched control subjects with normal renal function. Systemic exposure to abiraterone after a single oral 1 g dose did not increase in patients with end-stage renal disease on dialysis. Administration of abiraterone in patients with renal impairment including severe renal impairment does not require dose reduction (see Section 4.2 Dose and Method of Administration).
5.3 Preclinical Safety Data
Genotoxicity. Abiraterone acetate and abiraterone were devoid of genotoxic potential in the standard panel of genotoxicity tests including, an in vitro bacterial reverse mutation assay (the Ames test), an in vitro mammalian chromosome aberration test (using human lymphocytes) and an in vivo rat micronucleus assay. Genotoxicity studies have not been conducted with the main human metabolites of abiraterone.
Carcinogenicity. Carcinogenicity studies were not conducted with abiraterone acetate.
* All safety data from studies co-administering abiraterone with prednisone alone or prednisone/prednisolone have been included in the PI to ensure completeness of safety information. This safety information is applicable to coadministration of ARX-Abiraterone with prednisolone.
6 Pharmaceutical Particulars
6.1 List of Excipients
The tablets also contain the inactive ingredients:
250 mg tablet contains: lactose monohydrate; microcrystalline cellulose; croscarmellose sodium; povidone; sodium lauryl sulfate; magnesium stearate, silicon dioxide, macrogol 400, titanium dioxide and hypromellose.
500 mg tablet contains: lactose monohydrate, silicified microcrystalline cellulose croscarmellose sodium, sodium lauryl sulfate, magnesium stearate, hypromellose, colloidal anhydrous silica.
The tablet coating of the 500 mg tablet contains: Opadry II Purple 85F500067.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
ARX-Abiraterone 250 mg tablets. Store below 25°C.
ARX-Abiraterone 500 mg tablets. Store below 30°C.
6.5 Nature and Contents of Container
ARX-Abiraterone 250 mg tablets are provided in high density polyethylene white bottles fitted with a child-resistant polypropylene cap. A bottle contains 120 tablets.
ARX-Abiraterone 500 mg tablets are available in (PVC/PCTFE (Aclar)/Al blister packs of 60 tablets.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
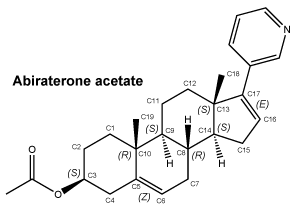
Molecular formula: C26H33NO2.
Molecular weight: 391.55.
CAS number. 154229-18-2.
7 Medicine Schedule (Poisons Standard)
S4 - Prescription Only Medicine.
Date of First Approval
06 August 2025
Date of Revision
06 August 2025
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.