Dexmedetomidine Viatris
Brand Information
| Brand name | Dexmedetomidine Viatris |
| Active ingredient | Dexmedetomidine |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Dexmedetomidine Viatris.
Summary CMI
DEXMEDETOMIDINE VIATRIS
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I being given DEXMEDETOMIDINE VIATRIS?
DEXMEDETOMIDINE VIATRIS contains the active ingredient dexmedetomidine hydrochloride. DEXMEDETOMIDINE VIATRIS can be used as a sedative (calming agent) if adults need to be calm or sleepy in the Intensive Care Unit whilst they are being ventilated (on a breathing machine). It can also be given to adults prior to an operation if they are not on a ventilator (breathing machine) and if they are required to be sleepy and calm for the procedure or surgery performed in a hospital.
For more information, see Section 1. Why am I being given DEXMEDETOMIDINE VIATRIS? in the full CMI.
2. What should I know before I am given DEXMEDETOMIDINE VIATRIS?
Do not use if you have ever had an allergic reaction to dexmedetomidine hydrochloride or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I am given DEXMEDETOMIDINE VIATRIS? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with DEXMEDETOMIDINE VIATRIS and affect how it works. These include medicines used to produce calmness or to help you sleep and strong pain relievers
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How is DEXMEDETOMIDINE VIATRIS given?
DEXMEDETOMIDINE VIATRIS is given by a slow injection (drip) into a vein by a doctor or nurse in the hospital. Your doctor will decide what dose you will receive. This depends on your condition and other factors such as your weight. The dose will be adjusted to keep you at the right depth of sleep or sedation.
More instructions can be found in Section 4. How is DEXMEDETOMIDINE VIATRIS given? in the full CMI.
5. What should I know while being given DEXMEDETOMIDINE VIATRIS?
| Things you should do |
|
| Driving or using machines |
|
For more information, see Section 5. What should I know while being given DEXMEDETOMIDINE VIATRIS? in the full CMI.
6. Are there any side effects?
Side effects of this medicine may include: headache; dizziness; light-headedness; nausea; vomiting; high temperature; feeling chills; dry mouth; constipation; diarrhoea; agitation; confusion; anxiety; hallucination; trouble with speech / understanding speech; fainting; tiredness; fluid retention (swelling in arms / legs); increased sweating; vision change; dry eyes; clammy skin; reduced or increased urine output; thirst; slowing or quickening of heart beat or palpitations; shortness of breath, rapid breathing or breathing difficulties; wheezing; tightness or pain in chest or arms that may spread to your neck, jaw or back; loud snoring, gasping for air during sleep; jaundice; unusual bruising; extreme fatigue; muscle twitching or spasms; restlessness; seizures.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
DEXMEDETOMIDINE VIATRIS
Active ingredient: Dexmedetomidine (as hydrochloride)
Consumer Medicine Information (CMI)
This leaflet provides important information about using DEXMEDETOMIDINE VIATRIS. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using DEXMEDETOMIDINE VIATRIS.
Where to find information in this leaflet:
1. Why am I being given DEXMEDETOMIDINE VIATRIS?
2. What should I know before I am given DEXMEDETOMIDINE VIATRIS?
3. What if I am taking other medicines?
4. How is DEXMEDETOMIDINE VIATRIS given?
5. What should I know while being given DEXMEDETOMIDINE VIATRIS?
6. Are there any side effects?
7. Product details
1. Why am I being given DEXMEDETOMIDINE VIATRIS?
DEXMEDETOMIDINE VIATRIS contains the active ingredient dexmedetomidine hydrochloride.
This medicine belongs to a group of medicines called alpha-2-receptor agonists. This medicine works by its actions on brain chemicals
DEXMEDETOMIDINE VIATRIS is used for:
Intensive Care Sedation
DEXMEDETOMIDINE VIATRIS can be used as a sedative (calming agent) if adults need to be calm or sleepy in the Intensive Care Unit of a hospital whilst they are being ventilated (on a breathing machine). It may be given as an infusion for up to 24 hours whilst in a hospital.
Procedural Sedation
DEXMEDETOMIDINE VIATRIS can be given to adults prior to an operation if they are not on a ventilator (breathing machine) and if they are required to be sleepy and calm for the procedure or surgery performed in a hospital.
Ask your doctor if you have any questions about why this medicine has been prescribed for you.
Your doctor may have prescribed it for another reason.
2. What should I know before I am given DEXMEDETOMIDINE VIATRIS?
Warnings
Do not use DEXMEDETOMIDINE VIATRIS if:
- you are allergic to dexmedetomidine hydrochloride or any of the ingredients listed at the end of this leaflet.
- Always check the ingredients to make sure you can use this medicine.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not give this medicine to a child under the age of 18 years.
Safety and effectiveness of DEXMEDETOMIDINE VIATRIS in children younger than 18 years have not been established.
Check with your doctor if you:
- have allergies to any other medicines, foods, preservatives or dyes.
- have any other medical conditions such as:
- diabetes
- kidney or liver problems
- heart problems
- high or low blood pressure - take any medicines for any other condition
- are over 65 years of age as elderly patients greater than 65 years old may be more prone to the blood pressure lowering effects of DEXMEDETOMIDINE VIATRIS.
An increased risk of death has been seen for patients 65 years of age and under when using this medicine. This increased risk is seen particularly in patients admitted to the intensive care unit for reasons other than after surgery; who have a more severe disease condition; and who are younger than 65. Your doctor will decide if this medicine is still suitable for you.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
It may affect your developing baby if you use it during pregnancy.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
The active ingredient in DEXMEDETOMIDINE VIATRIS passes into breast milk and there is a possibility that your baby may be affected.
If you have not told your doctor about any of the above, tell him/her before you are given DEXMEDETOMIDINE VIATRIS.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and DEXMEDETOMIDINE VIATRIS may interfere with each other.
These include:
- medicines used to produce calmness or to help you sleep such as sevoflurane, isoflurane, propofol, alfentanil and midazolam
- strong pain relievers
- medicines used to control blood pressure such as beta-blockers.
These medicines may be affected by DEXMEDETOMIDINE VIATRIS or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid if you are given this medicine whilst in hospital.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if they affect DEXMEDETOMIDINE VIATRIS.
4. How is DEXMEDETOMIDINE VIATRIS given?
How much is given
Your doctor will decide what dose you will receive. This depends on your condition and other factors such as your weight. The dose will be adjusted to keep you at the right depth of sleep or sedation.
How DEXMEDETOMIDINE VIATRIS is given
DEXMEDETOMIDINE VIATRIS is given by a slow injection (drip) into a vein. DEXMEDETOMIDINE VIATRIS should only be given by a doctor or nurse in the hospital.
If you use too much DEXMEDETOMIDINE VIATRIS
As DEXMEDETOMIDINE VIATRIS is given to you under the supervision of your doctor, it is very unlikely that you will receive too much.
Symptoms of an overdose may include extreme drowsiness, confusion, dizziness, weakness or becoming unconscious.
Your doctor has information on how to recognise and treat an overdose. Ask your doctor if you have any concerns.
If you think you or someone else have been given too much DEXMEDETOMIDINE VIATRIS, or if you experience any side effects, tell your doctor immediately.
5. What should I know while being given DEXMEDETOMIDINE VIATRIS?
Your doctor will be able to tell you whether there are any special instructions you should follow after you have been given DEXMEDETOMIDINE VIATRIS.
Things you should do
- Tell any other doctors, dentists, and pharmacists who treat you that you have been given this medicine.
- If you are going to have surgery, tell the surgeon or anaesthetist that you have been given this medicine.
It may affect other medicines used during surgery. - If you become pregnant while taking this medicine, tell your doctor immediately.
- If you are about to have any blood tests, tell your doctor that you have been given this medicine. It may interfere with the results of some tests
Driving or using machines
Be careful before you drive or use any machines or tools until you know how DEXMEDETOMIDINE VIATRIS affects you.
This medicine may cause dizziness, light-headedness, tiredness, drowsiness, and therefore affect alertness in some people. If you have any of these symptoms, do not drive, operate machinery or do anything else that could be dangerous.
Ask your doctor when you can return to work if it involves driving or operating machinery or heavy equipment.
Things to be careful of
If you feel light-headed, dizzy or faint when getting out of bed or standing up, get up slowly.
Standing up slowly, especially when you get up from bed or a chair, will help your body get used to the change in position and blood pressure. If this problem continues or gets worse, talk to your doctor.
Looking after your medicine
DEXMEDETOMIDINE VIATRIS will be stored in the hospital pharmacy or kept on the ward. The injection is kept in a cool dry place where the temperature stays below 25°C.
If it has expired or is damaged, it will be disposed of by the hospital pharmacist.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
If you are over 65 years of age you may have an increased chance of getting side effects.
This medicine helps provide sedation for most people, but it may have unwanted side effects in some people.
Do not be alarmed by the following list of side effects. You may not experience any of them.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
| Speak to your doctor if you have any of these less serious side effects and they worry you. They are usually mild and short-lived. |
Serious side effects
| Serious side effects | What to do |
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Some side effects can only be detected by blood or urine test. Your doctor will monitor your progress.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What DEXMEDETOMIDINE VIATRIS contains
| Active ingredient (main ingredient) | 118 microgram/mL of dexmedetomidine hydrochloride (equivalent to 100 microgram/mL dexmedetomidine base) |
| Other ingredients (inactive ingredients) | sodium chloride water for injections nitrogen |
Do not take this medicine if you are allergic to any of these ingredients.
What DEXMEDETOMIDINE VIATRIS looks like
DEXMEDETOMIDINE VIATRIS (200 microgram/2 mL - AUST R 271410) is a clear, colourless solution. It is available in 3 mL glass vials.
Who distributes DEXMEDETOMIDINE VIATRIS
Alphapharm Pty Ltd trading as Viatris
Level 1, 30 The Bond
30-34 Hickson Road
Millers Point NSW 2000
www.viatris.com.au
Phone: 1800 274 276
This leaflet was prepared in July 2025
DEXMEDETOMIDINE VIATRIS_cmi\Jul25/00
Brand Information
| Brand name | Dexmedetomidine Viatris |
| Active ingredient | Dexmedetomidine |
| Schedule | S4 |
MIMS Revision Date: 01 September 2025
1 Name of Medicine
Dexmedetomidine hydrochloride.
2 Qualitative and Quantitative Composition
Dexmedetomidine Viatris concentrated injection for intravenous infusion contains dexmedetomidine (as hydrochloride) 200 microgram/2 mL and is a sterile, non-pyrogenic, isotonic solution with a pH of 4.5 to 7.0.
Each 1 mL of Dexmedetomidine Viatris contains 118 micrograms of dexmedetomidine HCl (equivalent to 100 micrograms dexmedetomidine base) and 9 mg of sodium chloride in water. The solution is preservative-free and contains no additives or chemical stabilisers.
3 Pharmaceutical Form
Dexmedetomidine Viatris concentrated injection vial is supplied as a clear, colourless, isotonic solution free from visible particles. Dexmedetomidine Viatris is presented in a 3 mL vial and must be diluted prior to use.
4 Clinical Particulars
4.1 Therapeutic Indications
Intensive care unit (ICU) sedation. For sedation of initially intubated adult patients during treatment in an intensive care setting. The use of Dexmedetomidine Viatris by continuous infusion in these patients should not exceed 24 hours.
Procedural sedation. For sedation of non-intubated adult patients prior to and/or during surgical and other procedures.
4.2 Dose and Method of Administration
Note. Dexmedetomidine should be administered only by persons skilled in anaesthetics or in the management of patients in the intensive care setting. Due to the known pharmacological effects, patients should be continuously monitored.
Clinically significant events of bradycardia and sinus arrest have been associated with dexmedetomidine hydrochloride administration in young, healthy volunteers with high vagal tone or with different routes of administration including rapid intravenous or bolus administration of dexmedetomidine hydrochloride.
Dexmedetomidine should be individualised and titrated to the desired clinical effect.
ICU sedation. Initiation. For adult patients, Dexmedetomidine Viatris may be initiated with a loading infusion of 1 (one) microgram/kg over 10 to 20 minutes, if needed. The use of Dexmedetomidine Viatris by continuous infusion in these patients should not exceed 24 hours.
The use of loading dose of dexmedetomidine was associated with an increased rate of adverse event, including hypotension, hypertension and bradycardia, in clinical trials involving adult ICU patients.
For patients being converted from alternate sedative therapy a loading dose may not be required.
Maintenance of ICU sedation. Adult patients will generally require a maintenance infusion of 0.2 to 1 microgram/kg/h. The rate of the maintenance infusion should be adjusted to achieve the desired level of sedation. As a guide, it is recommended that 0.4 microgram/kg/h should be the initial maintenance infusion. If after approximately 5 minutes sedation is not adequate, the rate of infusion can be increased in increments of 0.1 microgram/kg/h or higher. Dosages as low as 0.05 microgram/kg/h have been used in clinical studies.
A dose reduction for both the loading and maintenance infusions should be considered in patients with impaired hepatic function and in patients over 65 years of age (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties).
Dexmedetomidine hydrochloride has been continuously infused in mechanically ventilated patients prior to extubation, during extubation, and post-extubation. It is not necessary to discontinue Dexmedetomidine Viatris prior to extubation.
Procedural sedation. Based on sedation scores, the loading infusion provides clinically effective onset of sedation 10 to 15 minutes after start of infusion.
Initiation. For adult patients, dexmedetomidine is generally initiated with a loading infusion of 1 (one) microgram/kg over 10 to 20 minutes for sedation of non-intubated patients undergoing surgical and other procedures, as well as, for initiation of awake fibre optic intubation.
For patients with impaired hepatic function and in patients over 65 years of age, the loading dose may be omitted or reduced, e.g. 0.5 microgram/kg over 10 minutes may be suitable.
For patients undergoing less invasive procedures, such as ophthalmic surgery, the loading dose may be reduced, e.g. 0.5 micrograms/kg over 10 minutes may be suitable.
Maintenance of procedural sedation. Following the loading dose, maintenance dosing of dexmedetomidine should generally be initiated at 0.6 microgram/kg/h and titrated to achieve desired clinical effect with doses ranging from 0.2 to 1 microgram/kg/h for all procedures. The rate of the maintenance infusion should be adjusted to achieve the targeted level of sedation.
Following the loading dose in awake fibre optic intubation, a fixed maintenance dose of 0.7 microgram/kg/h should be used until the endotracheal tube is secured.
A dose reduction should be considered in patients with impaired hepatic function and in patients over 65 years of age (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties).
Paediatric use. Safety and efficacy of dexmedetomidine hydrochloride has not been sufficiently established in paediatric patients (see Section 4.4 Special Warnings and Precautions for Use).
Administration. A controlled infusion device should be used to administer dexmedetomidine.
Strict aseptic technique must always be maintained during handling of dexmedetomidine infusion.
Preparation. Parenteral drug products should be inspected visually for particulate matter and discolouration prior to administration, whenever solution and container permit.
Dexmedetomidine Viatris must be diluted with 0.9% sodium chloride injection to achieve required concentration (4 microgram/mL) prior to administration. Preparation of infusion solutions is the same, whether for the loading dose or maintenance infusion.
To prepare the infusion, withdraw 2 mL of dexmedetomidine (as hydrochloride) concentrated injection and add to 48 mL of 0.9% sodium chloride to total 50 mL. Shake gently to mix well. Use as soon as practicable after dilution to reduce microbiological hazard. In-use stability has been demonstrated for 24 hours at 20 to 25°C. Parenteral products should be inspected visually for particulate matter and discolouration prior to administration.
Vials are intended for single use in one patient only. Discard any residue.
Compatibility. Dexmedetomidine has been shown to be compatible when administered with the following intravenous fluids: lactated Ringer's solution, 5% glucose in water, 0.9% sodium chloride in water, 20% mannitol in water.
Dexmedetomidine has been found to be compatible with water solutions of the following drugs when administered via Y-site injection: thiopental sodium, vecuronium bromide, pancuronium bromide, glycopyrronium bromide, phenylephrine hydrochloride.
4.3 Contraindications
Dexmedetomidine Viatris is contraindicated in patients with a known hypersensitivity to dexmedetomidine, or any of the excipients contained in Dexmedetomidine Viatris (see Section 6.1 List of Excipients).
4.4 Special Warnings and Precautions for Use
Drug administration. Dexmedetomidine hydrochloride is for hospital use only. Dexmedetomidine should be administered only by persons skilled in the management of patients in the intensive care or operating room setting. Due to the known pharmacological effects of dexmedetomidine hydrochloride, patients should be continuously monitored (MAC: Monitored Anaesthesia Care) for early signs of hypotension, hypertension, bradycardia, respiratory depression, airway obstruction, apnoea, dyspnoea and/or oxygen desaturation while receiving dexmedetomidine hydrochloride. Supplemental oxygen should be immediately available and provided when indicated.
Continuous electrocardiogram (ECG), blood pressure, and oxygen saturation monitoring are recommended during infusion of dexmedetomidine.
Dexmedetomidine may cause reduced lacrimation. Lubrication of the patient's eyes should be considered when administering dexmedetomidine to avoid corneal dryness.
Dexmedetomidine Viatris is only to be used for procedural sedation with the provision of appropriate monitoring and under the constant supervision of an appropriately trained medical practitioner. Although dexmedetomidine has sedative effects it has not been shown to be amnestic. Should amnesia be desired during procedural sedation then a drug with amnestic properties (such as a benzodiazepine) should be co-administered.
Hypotension, bradycardia and sinus arrest. Clinical events of bradycardia and sinus arrest have been associated with dexmedetomidine administration in young, healthy adult volunteers with high vagal tone or with different routes of administration including rapid intravenous or bolus administration of dexmedetomidine.
Reports of hypotension and bradycardia have been associated with dexmedetomidine infusion. Some of these cases have resulted in fatalities. If medical intervention is required, treatment may include decreasing or stopping the infusion of dexmedetomidine, increasing the rate of IV fluid administration, elevation of the lower extremities, and use of pressor agents. Because dexmedetomidine has the potential to augment bradycardia induced by vagal stimuli, clinicians should be prepared to intervene. The intravenous administration of anticholinergic agents (e.g. glycopyrronium bromide, atropine) should be considered to modify vagal tone. In clinical trials, glycopyrronium bromide or atropine were effective in the treatment of most episodes of dexmedetomidine induced bradycardia. However, in some patients with significant cardiovascular dysfunction, more advanced resuscitative measures were required.
Caution should be exercised when administering dexmedetomidine to patients with advanced heart block and/or severe ventricular dysfunction. Because dexmedetomidine decreases sympathetic nervous system activity, hypotension and/or bradycardia may be expected to be more pronounced in hypovolaemic patients and in those with diabetes mellitus, severe cardiac disease, chronic hypertension and in elderly patients.
In situations where other vasodilators or negative chronotropic agents are administered, coadministration of dexmedetomidine could have an additive pharmacodynamic effect and should be administered with caution.
Clinical events of bradycardia or hypotension may be potentiated when dexmedetomidine is used concurrently with propofol or midazolam. Therefore, consider a reduction in the dose of midazolam or propofol.
Elderly patients over 65 years of age, or diabetic patients, are more prone to hypotension with the administration of dexmedetomidine. All episodes either spontaneously reversed or were treated with standard therapy.
Transient hypertension. Transient hypertension has been observed primarily during the loading infusion, associated with initial peripheral vasoconstrictive effects of dexmedetomidine and relatively higher plasma concentrations achieved during the loading infusion. If intervention is necessary, reduction of the loading infusion rate may be considered. Following the loading infusion, the central effects of dexmedetomidine dominate and the blood pressure usually decreases.
Arousability. Patients receiving dexmedetomidine have been observed to be rousable and alert when stimulated. This is an expected component of dexmedetomidine sedation and should not be considered a lack of efficacy in the absence of other clinical signs and symptoms.
Withdrawal. Although not specifically studied, if dexmedetomidine is administered chronically and stopped abruptly, withdrawal symptoms similar to those reported for another alpha-2-adrenergic agent, clonidine, may result. These symptoms include nervousness, agitation, and headaches, accompanied or followed by a rapid rise in blood pressure and elevated catecholamine concentrations in the plasma. Dexmedetomidine Viatris should not be administered for greater than 24 hours.
Procedural sedation. In adult subjects, withdrawal symptoms were not seen after discontinuation of short-term infusions of dexmedetomidine (< 6 h).
Adrenal insufficiency. Dexmedetomidine had no effect on ACTH-stimulated cortisol release in dogs after a single dose; however, after the subcutaneous infusion of dexmedetomidine for one week, the cortisol response to ACTH was diminished by approximately 40%.
In a clinical study, prolonged infusions of dexmedetomidine at doses up to 1.4 microgram/kg/h were not associated with significant adrenocortical suppression.
Hyperthermia. Dexmedetomidine Viatris may induce hyperthermia that may be resistant to traditional cooling methods. Dexmedetomidine Viatris should be discontinued and hyperthermia should be managed with conventional medical measures.
Use in hepatic impairment. Since dexmedetomidine clearance decreases with increasing severity of hepatic impairment, dose reductions should be considered in patients with impaired hepatic function (see Section 4.2 Dose and Method of Administration).
Risk of mortality in ICU patients ≤ 65 years old. Use of dexmedetomidine greater than 24 hours has been associated with an increased mortality in critically ill adult ICU patients 65 years of age and younger compared to usual care (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Seizures. Dexmedetomidine lacks the anticonvulsant action of some other sedatives and so will not suppress underlying seizure activity.
Incompatibility. Dexmedetomidine has been shown to be incompatible when administered with the following drugs: amphotericin B, diazepam (see Section 6.2 Incompatibilities).
Use in the elderly. Dexmedetomidine is known to be substantially excreted by the kidney, and the risk of adverse reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection in elderly patients, and it may be useful to monitor renal function (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Paediatric use. The safety of dexmedetomidine in paediatric patients below 18 years of age has not been sufficiently established for procedural or ICU sedation. Therefore, Dexmedetomidine Viatris is not recommended in this population (see Section 4.2 Dose and Method of Administration; Section 5.1 Pharmacodynamic Properties, Clinical trials).
Effects on laboratory tests. No data available.
4.5 Interactions with Other Medicines and Other Forms of Interactions
Anaesthetics/sedatives/hypnotics/opioids. Co-administration of dexmedetomidine hydrochloride is likely to lead to an enhancement of effects with anaesthetics, sedatives, hypnotics, and opioids. Specific studies have confirmed these effects with sevoflurane, isoflurane, propofol, alfentanil, and midazolam. No pharmacokinetic interactions between dexmedetomidine and isoflurane, propofol, alfentanil, and midazolam were demonstrated. However, due to pharmacodynamic effects, when co-administered with dexmedetomidine hydrochloride, a reduction in dosage with these agents may be required.
Neuromuscular blockers. No clinically meaningful increases in the magnitude of neuromuscular blockade and no pharmacokinetic interactions were observed with dexmedetomidine hydrochloride and rocuronium administration.
Drugs with cardiovascular activities. The possibility of enhanced hypotensive and bradycardic effects should be considered in patients receiving other medicinal products causing these effects, for example beta blockers, although additional effects in an interaction study with esmolol were modest.
Cytochrome P450. In vitro studies did not demonstrate evidence for clinically relevant cytochrome P450 mediated drug interactions.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. Dexmedetomidine did not affect reproductive capacity or fertility in male or female rats after daily subcutaneous injections at doses up to 54 microgram/kg/day for 10 weeks prior to mating in males and 3 weeks prior to mating and during mating in females. Systemic exposure (AUC0-24h) at this dose level was less than anticipated at the maximum recommended human dose of 17.8 microgram/kg.
Use in pregnancy. (Category B1)
Radiolabelled dexmedetomidine administered subcutaneously to female rats on gestation day 18 crossed the placental barrier to fetal tissue.
Teratogenic effects were not observed following administration of dexmedetomidine at subcutaneous doses up to 200 microgram/kg/day in rats or IV doses up to 96 microgram/kg/day in rabbits. Systemic exposure (AUC1-24h) at these dose levels was 3 to 5 times greater than those in humans at the maximum recommended dose of 17.8 microgram/kg. In rats, fetal and pup body weights were reduced at SC doses ≥ 6 microgram/kg/day, postimplantation loss was increased at 200 microgram/kg/day, and perinatal mortality was increased at SC doses ≥ 18 microgram/kg/day. These findings are consistent with those of clonidine, another alpha2-adrenoreceptor agonist. Dexmedetomidine has no effect on fetal body weight or embryo fetal viability at IV doses as high as 96 microgram/kg/day in rabbits. Dexmedetomidine also produced delayed motor development in rat pups at a dose of 32 microgram/kg (less than the maximum recommended human intravenous dose). No such effects were observed at a dose of 2 microgram/kg.
There are no adequate and well-controlled studies in pregnant women. Dexmedetomidine has been shown to cross the placental barrier in human published studies. It has been reported that prenatal exposure to dexmedetomidine may be associated with some degree of functional impairment at birth in some neonates. Dexmedetomidine should be used during pregnancy only if the potential benefits justify the potential risk to the fetus.
Labour and delivery. The safety of dexmedetomidine in labour and delivery has not been studied and is, therefore, not recommended for obstetrics, including caesarean section deliveries. Perioperative administration of dexmedetomidine in pregnant women receiving general anaesthesia for elective caesarean section was associated with a longer time to clinical recovery and extubation compared with remifentanil.
Use in lactation. Dexmedetomidine is excreted in human milk, but no studies assessing the effects of dexmedetomidine in breastfed children and on milk production have been performed. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for dexmedetomidine and any potential adverse effects on the breastfed child from dexmedetomidine.
A lactating woman may consider interrupting breastfeeding and pumping and discarding breast milk for 24 hours after receiving dexmedetomidine in order to minimise potential drug exposure to a breastfed neonate. Radiolabelled dexmedetomidine administered subcutaneously to lactating female rats was distributed to but did not accumulate in milk.
4.7 Effects on Ability to Drive and Use Machines
Patients should be advised that performance of activities requiring mental alertness, such as operating a motor vehicle or hazardous machinery, may be impaired for some time after sedation.
4.8 Adverse Effects (Undesirable Effects)
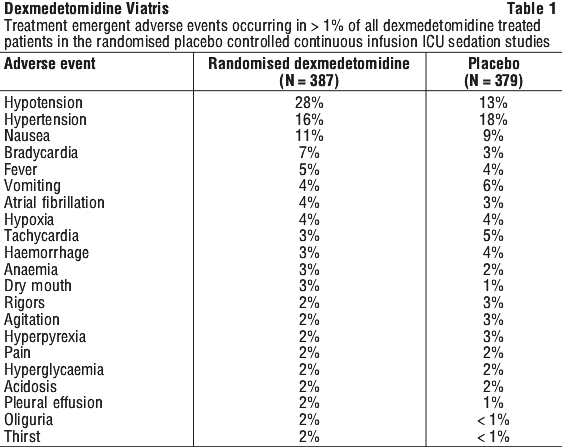
ICU sedation. Adverse event information derived from the placebo-controlled, continuous infusion trials of dexmedetomidine for sedation in the surgical ICU setting in which 387 patients received dexmedetomidine. In these studies, the mean total dose was 7.06 microgram/kg (SD = 2.86), mean dose per hour was 0.51 microgram/kg/h (SD = 0.39) and the mean duration of infusion of 15.6 hours (range: 0.17 to 29.08). The population was between 19 to 83 years of age, 43% over 65 years of age, 73% male and 97% Caucasian. Overall, the most frequently observed treatment-emergent adverse events included hypotension, hypertension, nausea, bradycardia, fever, vomiting, hypoxia, tachycardia and anaemia (see Table 1).
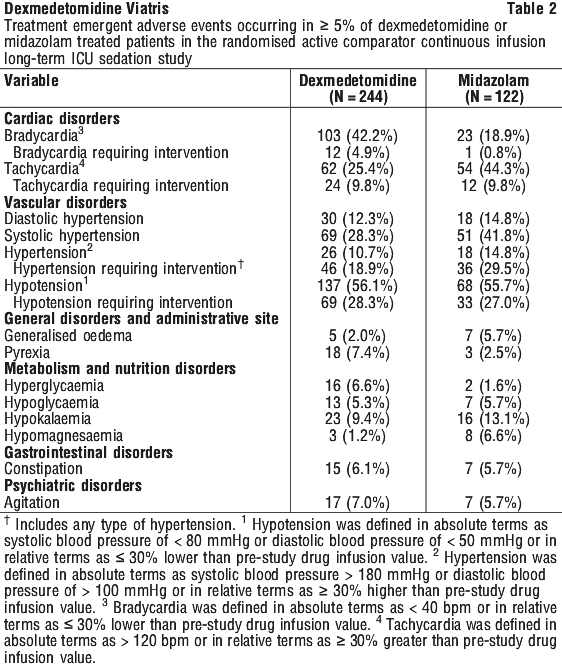
Procedural sedation. Adverse event information is derived from the two primary phase 3 trials for procedural sedation in which 318 patients received dexmedetomidine. The mean total dose was 1.6 microgram/kg (range: 0.5 to 6.7), mean dose per hour was 1.3 microgram/kg/h (range: 0.3 to 6.1) and the mean duration of infusion of 1.5 hours (range: 0.1 to 6.2). The population was between 18 to 93 years of age, 30% over 65 years of age, 52% male and 61% Caucasian.
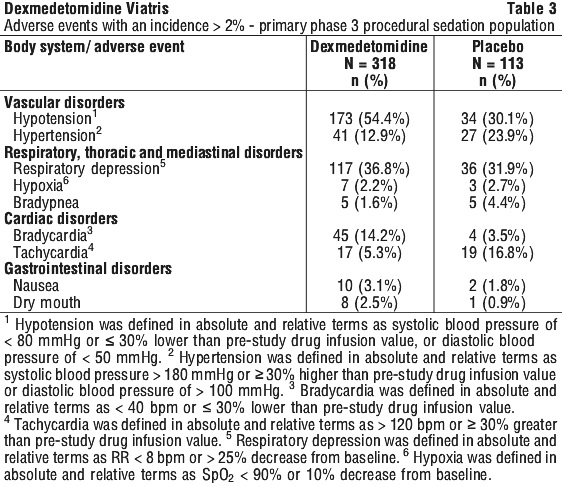
Treatment-emergent adverse events occurring at an incidence of > 2% are provided in Table 3. The majority of the adverse events were assessed as mild in severity. The most frequent adverse events were hypotension, bradycardia, and dry mouth. Pre-specified criteria for the vital signs to be reported as adverse events are footnoted below Table 3. Respiratory depression and hypoxia was similar in the dexmedetomidine and placebo groups when evaluated against the pre-specified criteria. The incidence of absolute respiratory depression and hypoxia was less in the dexmedetomidine-treated patients than the placebo patients (3.04% vs 12.7%) in the MAC trial.
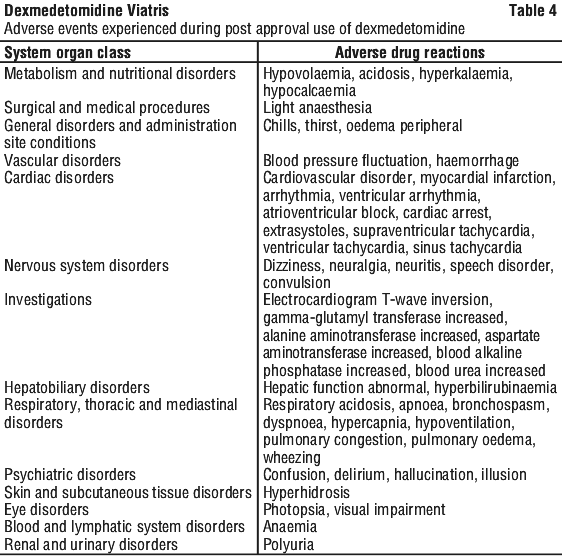
Hypotension and bradycardia were the most common adverse reactions associated with the use of dexmedetomidine during post approval use of the drug. Table 4 lists adverse drug reactions (ADRs within each standard system organ class (SOC).
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
4.9 Overdose
The tolerability of dexmedetomidine was noted in one study in which healthy adult subjects achieved plasma concentrations from 1.8 up to 13 times the upper boundary of the therapeutic range. The most notable effects observed in two subjects who achieved the highest plasma concentrations were 1st degree AV block and 2nd degree heart block. No haemodynamic compromise was noted with the AV block and the heart block resolved spontaneously within one minute.
Of five adult patients reported with overdose of dexmedetomidine in the phase II/III ICU sedation studies, two had no symptoms reported; one patient received a 2 microgram/kg loading dose over 10 minutes (twice the recommended loading dose) and one patient received a maintenance infusion of 0.8 microgram/kg/h. Two other patients who received a 2 microgram/kg loading dose over 10 minutes experienced bradycardia with or without hypotension. One patient, who received a loading bolus dose of undiluted (100 microgram/mL) dexmedetomidine (19.4 microgram/kg), had cardiac arrest from which he was successfully resuscitated.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: psycholeptics, other hypnotics and sedatives.
Mechanism of action. Dexmedetomidine is a relatively selective alpha2-adrenoreceptor agonist with sedative pharmacologic properties.
The sedative actions of dexmedetomidine are believed to be mediated primarily by post-synaptic alpha2-adrenoreceptors, which in turn act on inhibitory pertussis-toxin-sensitive G protein, thereby increasing conductance through potassium channels. The site of the sedative effects of dexmedetomidine has been attributed to the locus coeruleus. The analgesic actions are believed to be mediated by a similar mechanism of action at the brain and spinal cord level.
Alpha2-selectivity was observed in animals following slow intravenous (IV) infusion of low and medium doses (10-300 microgram/kg). Both alpha1 and alpha2 activity was observed following slow IV infusion of high doses (≥ 1000 microgram/kg) or with rapid IV administration. Dexmedetomidine has a low affinity for beta adrenergic, muscarinic, dopaminergic and serotonin receptors.
Clinical trials. ICU sedation. Two randomised, double-blind, parallel-group, placebo-controlled multicentre clinical trials included 754 adult patients being treated in a surgical intensive care unit (ICU). All patients were initially intubated and received mechanical ventilation.
These trials evaluated the sedative properties of dexmedetomidine by comparing the amount of rescue medication (midazolam in one trial and propofol in the second) required to achieve a specified level of sedation (using the standardised Ramsay Sedation Scale) between dexmedetomidine and placebo from onset of treatment to extubation or to a total treatment duration of 24 hours. The Ramsay level of sedation scale is displayed in Table 5.

A second prospective primary analysis assessed the sedative effects of dexmedetomidine by comparing the percentage of patients who achieved a Ramsay Sedation Score of ≥ 3 during intubation without the use of additional rescue medication. A significantly greater percentage of patients in the dexmedetomidine group maintained a Ramsay Sedation Score of ≥ 3 without receiving any midazolam rescue compared to the placebo group (see Table 6).
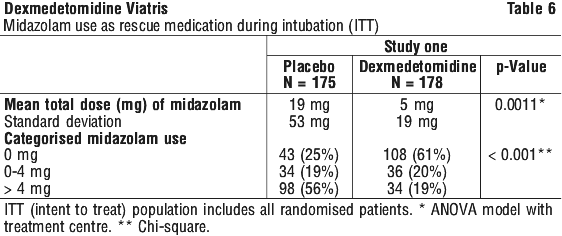
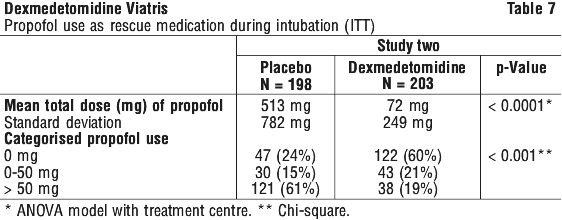
In the second study, 198 adult patients were randomised to receive placebo and 203 to receive dexmedetomidine by intravenous infusion at a dose of 0.4 microgram/kg/h (with allowed adjustment between 0.2 and 0.7 microgram/kg/h) following an initial loading infusion of 1 (one) microgram/kg IV over 10 minutes. The study drug infusion was adjusted to maintain a Ramsay Sedation Score of ≥ 3. Patients were allowed to receive "rescue" propofol as needed to augment the study drug infusion. In addition, morphine sulfate was administered as needed for pain. The primary outcome measure for this study was the total amount of rescue medication (propofol) needed to maintain sedation as specified while intubated.
Patients randomised to placebo received significantly more propofol than patients randomised to dexmedetomidine (see Table 7).
A significantly greater percentage of patients in the dexmedetomidine group compared to the placebo group maintained a Ramsay Sedation Score of ≥ 3 without receiving any propofol rescue (see Table 7).
Mortality in ICU patients ≤ 65 years old. In the SPICE III pragmatic randomised controlled trial of 3904 critically ill adult ICU patients dexmedetomidine was used as primary sedative and compared with usual care. There was no overall difference in 90-day mortality between the dexmedetomidine and usual care group (mortality 29.1% in both groups), but a heterogeneity of effect from age on mortality was observed. Dexmedetomidine was associated with an increased mortality in the age-group ≤ 65 years (odds ratio 1.26; 95% credibility interval 1.02 to 1.56) compared to alternative sedatives. While the mechanism is unclear, this heterogeneity of effect on mortality from age was most prominent in patients admitted for reasons other than post-operative care, and increased with increasing APACHE II scores and with decreasing age. These findings should be weighed against the expected clinical benefit of dexmedetomidine compared to alternative sedatives in younger patients.
In the published study, exposure to dexmedetomidine was greater than 24 hours with a median duration of treatment of 2.56 days (interquartile range, 1.10 to 5.23). The administration of dexmedetomidine was continued as clinically required for up to 28 days after randomisation (see Section 4.4 Special Warnings and Precautions for Use).
ICU sedation-elderly. A total of 729 patients in the clinical studies were 65 years of age and over. A total of 200 patients were 75 years of age and over. In patients greater than 65 years of age, a higher incidence of bradycardia and hypotension was observed following administration of dexmedetomidine (see Section 4.4 Special Warnings and Precautions for Use).
Consideration should be given to lower initial loading and maintenance doses in patients over 65 years of age and careful monitoring for the development of hypotension when up titrating the maintenance dose (see Section 4.2 Dose and Method of Administration).
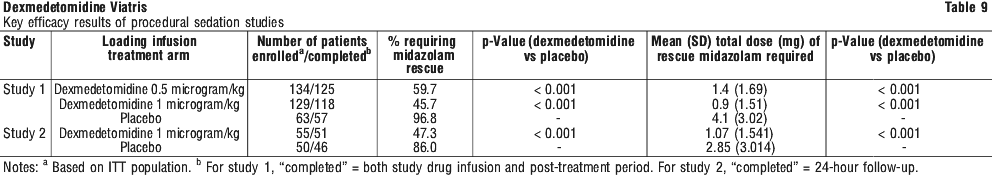
Procedural sedation. The safety and efficacy of dexmedetomidine for sedation of non-intubated patients prior to and/or during surgical and other procedures was evaluated in two randomised, double-blind, placebo-controlled multicentre clinical trials. Study 1 evaluated the sedative properties of dexmedetomidine in patients having a variety of elective surgeries/procedures performed under monitored anaesthesia care. Study 2 evaluated dexmedetomidine in patients undergoing awake fibreoptic intubation (AFOI) prior to a surgical or diagnostic procedure.
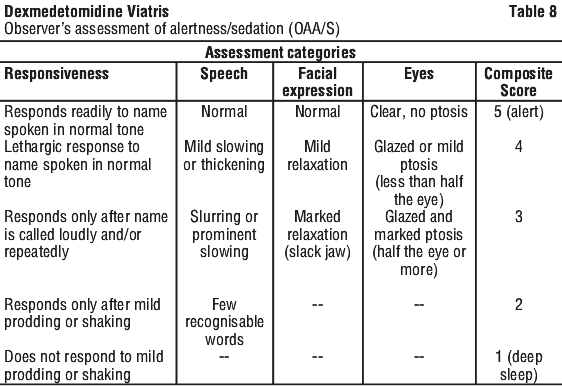
In study 1, the sedative properties of dexmedetomidine were evaluated by comparing the percent of patients not requiring rescue midazolam to achieve a specified level of sedation using the standardised observer's assessment of alertness/sedation scale between dexmedetomidine and placebo. The observer's assessment of alertness/sedation scale (see Table 8).
In study 2, the sedative properties of dexmedetomidine were evaluated by comparing the percent of patients requiring rescue midazolam to achieve or maintain a specified level of sedation using the Ramsay sedation scale score > 2 (see Table 2) during AFOI. Patients were randomised to receive a loading infusion of dexmedetomidine 1 microgram/kg or placebo (normal saline) given over 10 minutes and followed by a fixed maintenance infusion of 0.7 microgram/kg/h. After achieving the desired level of sedation, topicalisation of the airway occurred. Patients were allowed to receive rescue midazolam as needed to achieve and/or maintain an RSS score of > 2. Demographic characteristics were similar between the dexmedetomidine and placebo groups.
Paediatric studies. A US double-blind and two open-label studies in ICU sedation did not meet their primary efficacy endpoint, and the safety data were insufficient to fully characterise the safety profile of dexmedetomidine.
One open-label ICU sedation study conducted in Japanese patients did meet its primary efficacy endpoint.
The safety profile of dexmedetomidine in these studies was generally similar to that of adults, although increased frequencies of adverse events of bradycardia, hypotension, and respiratory depression were seen in the Japan ICU sedation study.
One open-label study conducted in paediatric patients for procedural sedation also did not meet its efficacy endpoint.
5.2 Pharmacokinetic Properties
Following intravenous administration, dexmedetomidine exhibits the following pharmacokinetic parameters: a rapid distribution phase with a distribution half-life (t1/2) of approximately six minutes; a terminal elimination half-life (t1/2) of approximately two hours; and steady-state volume of distribution (Vss) of approximately 118 litres. Clearance (CL) is estimated to be approximately 39 L/h. The mean body weight associated with this clearance estimate was 72 kg.
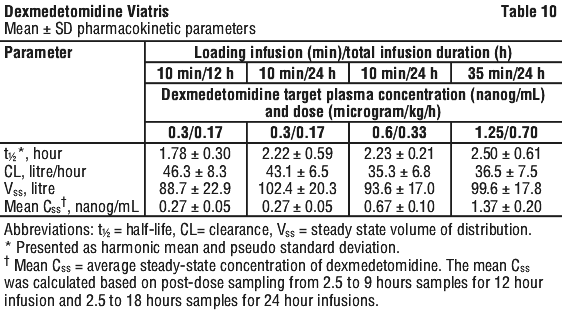
Dexmedetomidine exhibits linear kinetics in the dosage range of 0.2 to 0.7 microgram/kg/h when administered by IV infusion for up to 24 hours. Table 10 shows the main pharmacokinetic parameters when dexmedetomidine was infused (after appropriate loading doses) at maintenance infusion rates of 0.17 microgram/kg/h (target plasma concentration of 0.3 nanogram/mL) for 12 and 24 hours, 0.33 microgram/kg/h (target plasma concentration of 0.6 nanogram/mL) for 24 hours, and 0.70 microgram/kg/h (target plasma concentration of 1.25 nanogram/mL) for 24 hours.
Metabolism. Dexmedetomidine undergoes almost complete biotransformation with very little unchanged dexmedetomidine excreted in urine and faeces. Biotransformation involves both direct glucuronidation as well as cytochrome P450 mediated metabolism. The major metabolic pathways of dexmedetomidine are: direct N-glucuronidation to inactive metabolites; aliphatic hydroxylation (mediated primarily by CYP2A6 with a minor role of CYP1A2, CYP2E1, CYP2D6 and CYP2C19) of dexmedetomidine to generate 3-hydroxydexmedetomidine, the glucuronide of 3-hydroxydexmedetomidine, and 3-carboxydexmedetomidine; and N-methylation of dexmedetomidine to generate 3-hydroxy-N-methyldexmedetomidine, 3-carboxy-N-methyldexmedetomidine, and N-methyldexmedetomidine-O-glucuronide.
Excretion. The terminal elimination half-life (t1/2) of dexmedetomidine is approximately 2 hours and clearance is estimated to be approximately 39 L/h. A mass balance study demonstrated that after nine days an average of 95% of the radioactivity, following IV administration of radiolabelled dexmedetomidine, was recovered in the urine and 4% in the faeces. No unchanged dexmedetomidine was detected in the urine. Approximately 85% of the radioactivity recovered in the urine was excreted within 24 hours after the infusion. Fractionation of the radioactivity excreted in urine demonstrated that products of N-glucuronidation accounted for approximately 34% of the cumulative urinary excretion. In addition, aliphatic hydroxylation of parent drug to form 3-hydroxydexmedetomidine, the glucuronide of 3-hydroxydexmedetomidine, and 3-carboxylic acid-dexmedetomidine together represented approximately 14% of the dose in urine. N-methylation of dexmedetomidine to form 3-hydroxy-N-methyl dexmedetomidine, 3-carboxy-N-methyldexmedetomidine, and N-methyldexmedetomidine-O-glucuronide accounted for approximately 18% of the dose in urine. The N-methyl metabolite itself was a minor circulating component and was undetected in urine. Approximately 28% of the urinary metabolites have not been identified.
Special populations. Patients with hepatic impairment. In subjects with varying degrees of hepatic impairment (Child-Pugh class A, B, or C), clearance values for dexmedetomidine hydrochloride were lower than in healthy subjects. The mean clearance values for patients with mild, moderate, and severe hepatic impairment were 74%, 64% and 53%, of those observed in the normal healthy subjects, respectively. Mean clearances for free drug were 59%, 51% and 32% of those observed in the normal healthy subjects, respectively.
Although dexmedetomidine hydrochloride is dosed to effect, it may be necessary to consider dose reduction depending on the degree of hepatic impairment (see Section 4.2 Dose and Method of Administration).
Patients with renal impairment. Dexmedetomidine hydrochloride pharmacokinetics (Cmax, Tmax, AUC, t1/2, CL, and Vss) were not significantly different in patients with severe renal impairment (creatinine clearance: < 30 mL/min) compared to healthy subjects.
In view of the limited toxicological data and the potential for higher plasma metabolite concentrations in patients with severe renal impairment, caution is advised with prolonged dosing in such patients.
Male and female patients. There was no observed difference in dexmedetomidine hydrochloride pharmacokinetics due to gender.
Elderly. The pharmacokinetic profile of dexmedetomidine hydrochloride was not altered by age. However, as with many drugs, the elderly may be more sensitive to the effects of dexmedetomidine. In clinical trials, there was a higher incidence of bradycardia and hypotension in elderly patients.
Children. The pharmacokinetic profile of dexmedetomidine hydrochloride has not been studied in children.
5.3 Preclinical Safety Data
Genotoxicity. Dexmedetomidine was not mutagenic in vitro, in either the bacterial reverse mutation assay (E. coli and Salmonella typhimurium) or the mammalian cell forward mutation assay (mouse lymphoma). In a mouse micronucleus study, dexmedetomidine was not cytotoxic to bone marrow and did not increase the numbers of micronucleated PCEs at any dose tested, both in animals maintained at room temperature and in those kept warm. In addition, dexmedetomidine did not induce chromosomal aberrations in cultured human peripheral blood lymphocytes in the absence or presence of an exogenous metabolic activation system comprised of a human S9 homogenate.
Carcinogenicity. Animal carcinogenicity studies have not been performed with dexmedetomidine.
6 Pharmaceutical Particulars
6.1 List of Excipients
Sodium chloride, water for injection, nitrogen.
6.2 Incompatibilities
Compatibility of dexmedetomidine hydrochloride with co-administration of blood, serum, or plasma has not been established. Dexmedetomidine Viatris must not be mixed with other medicinal products.
Dexmedetomidine hydrochloride has been shown to be incompatible when administered with the following drugs: amphotericin B, diazepam (see Section 4.4 Special Warnings and Precautions for Use).
Compatibility studies have demonstrated the potential for absorption of dexmedetomidine to some types of natural rubber.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
Use as soon as practicable after dilution to reduce microbiological hazard. In-use stability has been demonstrated for 24 hours at 20 to 25°C.
6.4 Special Precautions for Storage
Store below 25°C.
See Section 6.3 Shelf Life for storage directions after dilution.
6.5 Nature and Contents of Container
Dexmedetomidine Viatris, (as dexmedetomidine hydrochloride), 200 microgram/2 mL concentrated injection vials are available in:
Container type: 3 mL vials (glass type I clear).
Pack sizes: Cartons of 1, 5 or 25 vials.
Some pack sizes may not be marketed.
Australian register of therapeutic goods (ARTG). AUST R 271410 - Dexmedetomidine Viatris dexmedetomidine (as hydrochloride) 200 microgram/2 mL concentrated injection vial.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking it to your local pharmacy.
6.7 Physicochemical Properties
Dexmedetomidine hydrochloride is a white or almost white powder, freely soluble in water and its pKa is 7.1. The partition coefficient in octanol: water at pH 7.84 is 2.89.
Chemical structure. Chemical name: (+)-4-(S)-[1-(2,3-dimethylphenyl) ethyl]-1H-imidazole monohydrochloride.
Structural formula:
CAS number. CAS-145108-58-3.
7 Medicine Schedule (Poisons Standard)
S4 (Prescription Only Medicine).
Date of First Approval
15 October 2018
Date of Revision
24 July 2025
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.