Entecavir Viatris
Brand Information
| Brand name | Entecavir Viatris |
| Active ingredient | Entecavir |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Entecavir Viatris.
Summary CMI
ENTECAVIR VIATRIS
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about taking this medicine, speak to your doctor or pharmacist.
1. Why am I taking ENTECAVIR VIATRIS?
ENTECAVIR VIATRIS contains the active ingredient entecavir (as monohydrate). ENTECAVIR VIATRIS is used to treat adults infected with hepatitis B virus.
For more information, see Section 1. Why am I taking ENTECAVIR VIATRIS? in the full CMI.
2. What should I know before I take ENTECAVIR VIATRIS?
Do not take if you have ever had an allergic reaction to ENTECAVIR VIATRIS or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I take ENTECAVIR VIATRIS? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with ENTECAVIR VIATRIS and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I take ENTECAVIR VIATRIS?
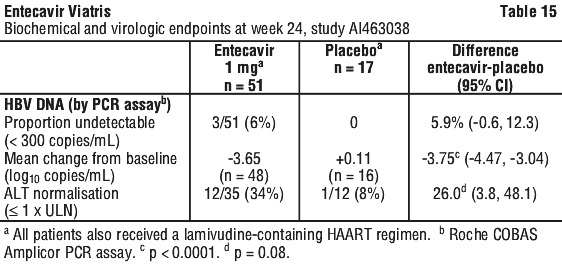
- The usual dose of ENTECAVIR VIATRIS is 0.5 mg or 1 mg once a day on an empty stomach.
- Swallow the tablets whole with a full glass of water.
More instructions can be found in Section 4. How do I take ENTECAVIR VIATRIS? in the full CMI.
5. What should I know while taking ENTECAVIR VIATRIS?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while taking ENTECAVIR VIATRIS? in the full CMI.
6. Are there any side effects?
Less serious side effects: diarrhoea; indigestion; tiredness; headache. Serious side effects: skin and the white part of eyes turns yellow (jaundice); urine turns dark; bowel movement (stool) turn light in colour; loss of appetite; nausea; lower stomach pain; lactic acidosis; serious liver problems; feeling cold (especially in arms and legs); feeling dizzy or light-headed; chills; fever; fast heartbeat; wheezing or coughing; difficulty breathing; dizziness; flushing; sweating and swelling of the face, tongue or other parts of the body. For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
ENTECAVIR VIATRIS
Active ingredient(s): Entecavir (as monohydrate)
Consumer Medicine Information (CMI)
This leaflet provides important information about taking ENTECAVIR VIATRIS. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about taking ENTECAVIR VIATRIS.
Where to find information in this leaflet:
1. Why am I taking ENTECAVIR VIATRIS?
2. What should I know before I take ENTECAVIR VIATRIS?
3. What if I am taking other medicines?
4. How do I take ENTECAVIR VIATRIS?
5. What should I know while taking ENTECAVIR VIATRIS?
6. Are there any side effects?
7. Product details
1. Why am I taking ENTECAVIR VIATRIS?
ENTECAVIR VIATRIS contains the active ingredient entecavir (as monohydrate). ENTECAVIR VIATRIS belongs to a group of medicines called antiviral medicines.
ENTECAVIR VIATRIS is used to treat adults infected with hepatitis B virus.
Infection by hepatitis B virus can lead to damage to the liver. ENTECAVIR VIATRIS reduces the amount of virus in your body and has been shown to improve the condition of the liver.
It is not known how safe ENTECAVIR VIATRIS is when taken for long periods.
Ask your doctor if you have any questions about why ENTECAVIR VIATRIS has been prescribed for you.
Your doctor may have prescribed it for another reason.
ENTECAVIR VIATRIS is not addictive.
2. What should I know before I take ENTECAVIR VIATRIS?
Warnings
Do not take ENTECAVIR VIATRIS if:
- you have an allergy to:
- any medicine containing entecavir monohydrate
- any of the ingredients listed at the end of this leaflet
Some of the symptoms of an allergic reaction may include:
- chills, fever
- fast heartbeat
- wheezing or coughing
- difficulty breathing
- dizziness
- flushing
- sweating and swelling of the face, lips, tongue or other parts of the body
Always check the ingredients to make sure you can take this medicine. - the expiry date printed on the pack has passed or if the packaging is torn or shows signs of tampering.
If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Check with your doctor if you:
- have allergies to any other medicines, foods, preservatives or dyes
- have or have had any of the following medical conditions:
- currently experience or have experienced any medical conditions especially any problems with your kidneys
- have HIV (human immunodeficiency virus) and you are not currently on HIV treatment
ENTECAVIR VIATRIS is not recommended in patients who have both HIV and hepatitis B and who are not currently receiving anti-HIV treatment. ENTECAVIR VIATRIS may affect your HIV virus which could impact on future treatment options for HIV.
- are lactose intolerant. ENTECAVIR VIATRIS contains lactose. - take any medicines for any other condition.
If you have not told your doctor about any of the above, tell him/her before you start taking ENTECAVIR VIATRIS.
It is important to remain under the care of your doctor during ENTECAVIR VIATRIS therapy and after stopping ENTECAVIR VIATRIS. You should report any new symptoms, medication or any other aspects affecting your health to your doctor. Your hepatitis B virus infection may get worse if you stop taking ENTECAVIR VIATRIS. If your doctor advises you to stop ENTECAVIR VIATRIS, they will monitor your health and perform regular blood tests to monitor your liver.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Tell your doctor if you are pregnant or plan to become pregnant.
Experience is limited with the use of ENTECAVIR VIATRIS in pregnant women. Therefore, it should not be used during pregnancy unless it is clearly needed. If there is an urgent need to consider ENTECAVIR VIATRIS during pregnancy, your doctor will discuss with you the risks and benefits involved. If you take ENTECAVIR VIATRIS while you are pregnant, talk to your doctor about how you can take part in the entecavir Pregnancy Registry. The purpose of the Pregnancy Registry is to collect information about the health of you and your baby.
Tell your doctor if you are breastfeeding or plan to breastfeed.
It is not known whether ENTECAVIR VIATRIS passes into breast milk. Therefore, to avoid possible side effects in the nursing infant, mothers should stop breast-feeding if they are taking ENTECAVIR VIATRIS.
Children
This medicine is not recommended for use in children under 16 years, as there have been no studies of its effects in children.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect ENTECAVIR VIATRIS.
4. How do I take ENTECAVIR VIATRIS?
How much to take
- The usual dose of ENTECAVIR VIATRIS is 0.5 mg or 1 mg once a day.
- If you have a medical problem with your kidneys your doctor may need to change how often you take your ENTECAVIR VIATRIS tablets.
- Your doctor will tell you what dose to take and how often you should take your ENTECAVIR VIATRIS tablets.
When to take ENTECAVIR VIATRIS
- Take your medicine any time of day provided it is on an empty stomach. Empty stomach means at least 2 hours before food or 2 hours after food.
- Talk to your doctor or pharmacist to work out when it is best for you to take your dose of ENTECAVIR VIATRIS.
How to take ENTECAVIR VIATRIS
- Swallow the tablets whole with a full glass of water on an empty stomach.
How long to take ENTECAVIR VIATRIS
- Continue taking your medicine for as long as your doctor tells you to.
This medicine helps to control your condition, but does not cure it. It is important to keep taking your medicine even if you feel well and for as long as your doctor tells you to. - Your doctor has prescribed ENTECAVIR VIATRIS to prevent hepatitis B virus from further damaging your liver.
ENTECAVIR VIATRIS is a very important treatment that can improve the inflammation and scar tissue caused by the hepatitis B virus in your liver and may reduce the chance of developing cirrhosis, liver failure and liver cancer.
It is important to take ENTECAVIR VIATRIS every day or as directed by your doctor, to not miss doses, and to make sure you have enough supply until you next see your doctor. - Do not stop taking ENTECAVIR VIATRIS or change the dose unless asked to do so by your doctor, even if you feel better, as it can be very dangerous.
It is extremely important that you do not stop without discussing with your doctor because if ENTECAVIR VIATRIS is suddenly stopped, the hepatitis B virus can become very active again and lead to sudden development of severe liver failure.
There is high risk of dying if liver failure develops and liver transplantation may be necessary to save your life.
If you forget to take ENTECAVIR VIATRIS
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take it as soon as you remember, and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for the dose you missed.
This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much ENTECAVIR VIATRIS
If you think that you or anyone else has taken too much ENTECAVIR VIATRIS, urgent medical attention may be needed.
You should immediately:
- phone the Poisons Information Centre
(by calling telephone 13 11 26) for advice, or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while taking ENTECAVIR VIATRIS?
Things you should do
- If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking ENTECAVIR VIATRIS.
- Tell any other doctors, dentists and pharmacists who treat you that you are taking ENTECAVIR VIATRIS.
- If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine.
It may affect other medicines used during surgery. - If you become pregnant while taking this medicine, tell your doctor immediately.
- If you are about to have any blood tests, tell your doctor that you are taking this medicine.
It may interfere with the results of some tests. - Keep all of your doctor's appointments so that your progress can be checked.
Things you should not do
- Do not take ENTECAVIR VIATRIS to treat any other complaints unless your doctor tells you to.
- Do not give your medicine to anyone else, even if they have the same condition as you.
- Do not stop taking your medicine or change the dosage without checking with your doctor.
If you stop taking it suddenly, your condition may worsen.
Things to be careful of
- If you feel light-headed, dizzy or faint when getting out of bed or standing up, get up slowly.
Standing up slowly, especially when you get up from bed or chairs, will help your body get used to the change in position and blood pressure. If this problem continues or gets worse, talk to your doctor. - Make sure that you visit your doctor regularly throughout your entire course of treatment with ENTECAVIR VIATRIS.
- When your treatment with ENTECAVIR VIATRIS is stopped, your doctor will continue to monitor you and take blood tests for several months.
- There is no evidence that ENTECAVIR VIATRIS reduces the risk of infecting others with hepatitis B through sexual contact or body fluids (including blood contamination).
Therefore, it is important to take appropriate precautions to prevent others being infected with hepatitis B. - Talk to your doctor about safe sexual practices that protect your partner. Never share needles. Do not share personal items that can have blood or bodily fluids on them, like toothbrushes and razor blades. A vaccine is available to protect those at risk of becoming infected with hepatitis B.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how ENTECAVIR VIATRIS affects you.
ENTECAVIR VIATRIS may cause dizziness in some people. It is not known if this was caused by ENTECAVIR VIATRIS. If you have this symptom, do not drive, operate machinery or do anything else that could be dangerous.
Looking after your medicine
- Keep your tablets in the blister pack until it is time to take them.
If you take the tablets out of the pack, they may not keep well. - Store your tablets below 25°C.
Follow the instructions on the carton on how to take care of your medicine properly.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Heat and dampness can destroy some medicines.
Keep it where young children cannot reach it.
A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Getting rid of any unwanted medicine
If you no longer need to take this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not take this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Do not be alarmed by the following list of side effects.
You may not experience any of them.
Less serious side effects
| Less serious side effects | What to do |
| Speak to your doctor or pharmacist if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
Signs or symptoms of liver problems:
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these very serious side effects. |
Lactic acidosis: It is a serious medical emergency that can cause death. Lactic acidosis must be treated in the hospital. Reports of lactic acidosis with ENTECAVIR VIATRIS generally involved patients who were seriously ill due to their liver disease or other medical condition.
Liver problems: Some people who have taken medicines like ENTECAVIR VIATRIS have developed serious liver problems called hepatotoxicity, with liver enlargement (hepatomegaly) and fat in the liver (steatosis). Hepatomegaly with steatosis is a serious medical emergency that can cause death.
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What ENTECAVIR VIATRIS contains
| Active ingredient (main ingredient) | entecavir (as monohydrate) |
| Other ingredients (inactive ingredients) |
|
| Potential allergens | Sugars as lactose |
Do not take this medicine if you are allergic to any of these ingredients.
What ENTECAVIR VIATRIS looks like
- ENTECAVIR VIATRIS 0.5 mg: A white, film-coated, round, biconvex, beveled edge tablet debossed with 'M' on one side of the tablet and 'EV1' on the other side (AUST R 220090).
Blister pack of 30 tablets. - ENTECAVIR VIATRIS 1 mg: A white, film-coated, round, biconvex, beveled edge tablet debossed with 'M' on one side of the tablet and 'EV2' on the other side (AUST R 220091).
Blister pack of 30 tablets.
Who distributes ENTECAVIR VIATRIS
Alphapharm Pty Ltd trading as Viatris
Level 1, 30 The Bond
30-34 Hickson Road
Millers Point NSW 2000
www.viatris.com.au
Phone: 1800 274 276
This leaflet was prepared in November 2025.
ENTECAVIR VIATRIS_cmi\Nov25/00
Brand Information
| Brand name | Entecavir Viatris |
| Active ingredient | Entecavir |
| Schedule | S4 |
MIMS Revision Date: 01 December 2022
1 Name of Medicine
Entecavir (as monohydrate).
2 Qualitative and Quantitative Composition
Each Entecavir Viatris tablet contains 0.5 mg or 1 mg of entecavir (as monohydrate).
Excipients with known effects. Sugars (as lactose).
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Entecavir Viatris 0.5 mg. A white, film-coated, round, biconvex, beveled edge tablet debossed with 'M' on one side of the tablet and 'EV1' on the other side.
Entecavir Viatris 1 mg. A white, film-coated, round, biconvex, beveled edge tablet debossed with 'M' on one side of the tablet and 'EV2' on the other side.
4 Clinical Particulars
4.1 Therapeutic Indications
Entecavir is indicated for the treatment of chronic hepatitis B virus infection in adults 16 years or older with evidence of active liver inflammation.
This indication is based on histologic, virologic, biochemical and serological responses in nucleoside treatment naïve and lamivudine resistant adult patients with HBeAg positive or HBeAg negative chronic HBV infection with compensated liver disease.
4.2 Dose and Method of Administration
Recommended dosage. Entecavir should be taken orally, on an empty stomach (at least 2 hours after a meal and at least 2 hours before the next meal).
The recommended oral dose of entecavir in adults and adolescents older than 16 years is 0.5 mg once daily. For lamivudine refractory patients [history of hepatitis B viremia while receiving lamivudine therapy or known lamivudine resistance (LVDR commonly called YMDD mutations)], the recommended dose is 1 mg once daily.
Renal impairment. In patients with renal impairment, the apparent oral clearance of entecavir decreased as creatinine clearance decreased (see Section 5.2 Pharmacokinetic Properties, Special populations). Dosage adjustment of entecavir is recommended for patients with creatinine clearance < 50 mL/min, including patients on haemodialysis or CAPD, as shown in Table 1.
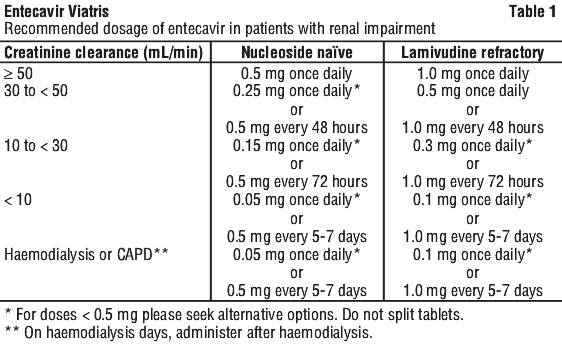
Duration of therapy. The optimal duration of treatment with entecavir for patients with chronic hepatitis B infection and the relationship between treatment and long-term outcomes such as cirrhosis and hepatocellular carcinoma are unknown.
4.3 Contraindications
Entecavir is contraindicated in patients with previously demonstrated hypersensitivity to entecavir or any component of the product. See Section 6.1 List of Excipients.
4.4 Special Warnings and Precautions for Use
Coinfection with HIV. Therapy with entecavir is not recommended for HIV/HBV coinfected patients who are not receiving highly active antiretroviral therapy. There is a potential for the development of HIV resistance if entecavir is used to treat chronic hepatitis B infection in patients with untreated HIV infection.
Lactic acidosis. Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues alone or in combination with antiretrovirals.
Exacerbations of hepatitis after discontinuation of treatment. Acute exacerbation of hepatitis has been reported in patients who have discontinued hepatitis B therapy, including therapy with entecavir (see Section 4.8 Adverse Effects (Undesirable Effects)). The majority of post-treatment exacerbations appear to be self limited. However, severe exacerbations, including fatalities, may occur. Hepatic function should be monitored closely for at least several months after discontinuation. If appropriate, resumption of hepatitis B therapy may be warranted.
Use in renal impairment. Dosage adjustment of entecavir is recommended for patients with renal impairment who have creatinine clearance < 50 mL/min (see Section 4.2 Dose and Method of Administration).
Liver transplant recipients. Limited data are available on the safety and efficacy of entecavir in liver transplant recipients. In a single arm, open label study, patients who had HBV DNA less than 172 IU/mL at the time of transplant were treated with entecavir 1 mg once daily post-transplant. On treatment, 15 subjects (23%) had liver related adverse events of interest: fourteen subjects had ascites (22%) and 1 subject each had bacterial peritonitis, hepatic encephalopathy, and recurrent HCC. In 12 of the 15 subjects, all liver related events occurred during the first 30 days post-transplant, and were considered postoperative complications. During the first 30 days post-transplant, 18 of 65 treated subjects (28%) or 8 of 61 evaluable subjects (13%) had episodes of acute liver rejection with 1 subject who required retransplantation. None of the 61 evaluable patients had virologic recurrence. The frequency and nature of adverse events and acute liver rejection in this study were consistent with those expected in patients who have received a liver transplant and the known safety profile of entecavir. (See Section 5.1 Pharmacodynamic Properties, Clinical trials).
Renal function should be carefully monitored before and during entecavir therapy in liver transplant recipients receiving an immunosuppressant that may affect renal function such as ciclosporine or tacrolimus (see Section 4.2 Dose and Method of Administration, Hepatic impairment; Section 5.2 Pharmacokinetic Properties, Hepatic impairment and Postliver transplant).
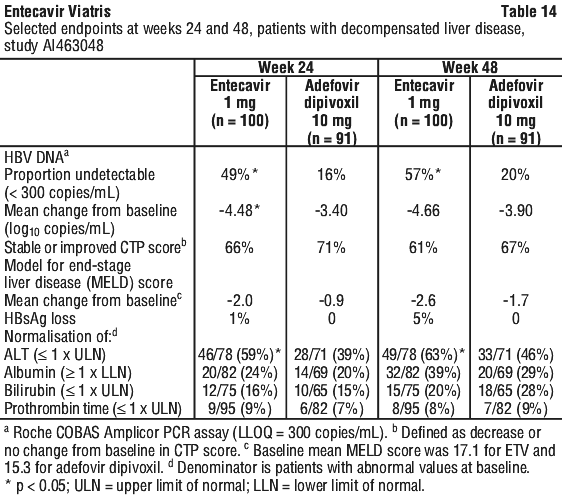
Decompensated liver disease. A study of entecavir at a dose of 1 mg once daily has been conducted in patients with decompensated liver disease (see Section 5.1 Pharmacodynamic Properties, Clinical trials; Section 4.8 Adverse Effects (Undesirable Effects)).
Coinfection with hepatitis C or D. There are no data on the efficacy of entecavir in patients coinfected with hepatitis C or D.
Paediatric use. Safety and effectiveness of entecavir in paediatric patients below the age of 16 years have not been established.
Use in the elderly. Clinical studies of entecavir did not include sufficient numbers of participants aged 65 years and over to determine whether they respond differently from younger participants. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. Entecavir is substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function (see Section 4.2 Dose and Method of Administration, Renal impairment).
Lactose. This medicinal product contains 62.5 mg of lactose monohydrate in each 0.5 mg daily dose and 125 mg of lactose monohydrate in each 1 mg daily dose.
Patients with rare hereditary problems of galactose intolerance, the Lapp lactase deficiency of glucose galactose malabsorption should not take this medicine.
Effects on laboratory tests. No data available.
Patient information. A consumer medicine information leaflet for Entecavir Viatris is available for patient information.
Patients should remain under the care of a physician while taking entecavir. They should discuss any new symptoms or concurrent medications with their physician.
Patients should be advised to take entecavir on an empty stomach (at least 2 hours before and at least 2 hours after a meal).
Patients should be informed that deterioration of liver disease may occur in some cases if treatment is discontinued, and that they should discuss any change in regimen with their physician.
Patients should be advised that treatment with entecavir has not been shown to reduce the risk of transmission of HBV to others through sexual contact or blood contamination.
4.5 Interactions with Other Medicines and Other Forms of Interactions
Medicinal products. Since entecavir is predominantly eliminated by the kidney (see Section 5.2 Pharmacokinetic Properties, Excretion), coadministration with medicinal products that reduce renal function or compete for active tubular secretion may increase serum concentrations of either medicinal product. Coadministration of entecavir with either lamivudine, adefovir dipivoxil or tenofovir disoproxil fumarate resulted in no significant drug interactions. The effects of coadministration of entecavir with other medicinal products that are excreted renally or affect renal function have not been evaluated. Patients should be monitored closely for adverse events when entecavir is coadministered with such medicinal products.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. Male and female rat fertility was unaffected by drug exposures (AUC) up to approximately 160 (male) and 230 (female) times that in humans treated with a daily dose of 1 mg. Testicular seminiferous tubule degeneration or germinal epithelial maturation arrest was observed in long-term rodent studies, at drug exposures that were ≥ 10 (mice) and 29 (rats) times the human value, and in dog studies at exposures > 379 times the human value. No testicular changes were evident in monkeys at exposures up to 114 times the clinical exposure.
Use in pregnancy. (Category B3)
Category B3: Drugs which have been taken by only a limited number of pregnant women and women of childbearing age, without an increase in the frequency of malformation or other direct or indirect harmful effects on the human fetus having been observed. Studies in animals have shown evidence of an increased occurrence of fetal damage, the significance of which is considered uncertain in humans.
There are no adequate and well controlled studies in pregnant women. Entecavir should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
There are no data on the effect of entecavir on transmission of HBV from mother to infant. Therefore, appropriate interventions should be used to prevent neonatal acquisition of HBV.
In animal experiments, embryofetal development was unaffected at drug exposures (AUC) of up to 23 (rats) and 175 (rabbits) times the maximum human exposure (1 mg daily dose). Slightly increased resorptions occurred in rats at a maternotoxic dose resulting in a higher drug exposure (150 times the human value), while embryofetal development was severely retarded at a maternotoxic dose resulting in a drug exposure > 2500 times the human value; tail and vertebral malformations were observed. A drug exposure 730 times the maximum human value resulted in increased resorptions and incomplete fetal hyoid ossification in rabbits, in the absence of maternal toxicity. Rat offspring development was unaffected at drug exposures approximately 230 times the human value, when mothers were treated from early gestation to the end of lactation.
Pregnancy registry. To monitor maternal fetal outcomes of pregnant women exposed to entecavir, a pregnancy registry has been established. Physicians are encouraged to register patients by calling 1-800-067-567.
Use in lactation. Entecavir and/or its conjugate metabolites are excreted in the milk of rats. It is not known whether excretion occurs in human milk. Mothers should be instructed not to breastfeed if they are taking entecavir.
4.7 Effects on Ability to Drive and Use Machines
The effects of this medicine on a person's ability to drive and use machines were not assessed as part of its registration.
4.8 Adverse Effects (Undesirable Effects)
Assessment of adverse reactions is based on four clinical studies in which 1720 patients with chronic HBV infection received double blind treatment with entecavir 0.5 mg/day (n = 679), entecavir 1 mg/day (n = 183), or lamivudine (n = 858) for up to 107 weeks. The safety profiles of entecavir and lamivudine were comparable in these studies. Among entecavir treated patients, the most common adverse events of any severity with at least a possible relation to entecavir were headache (9%), fatigue (6%), dizziness (4%) and nausea (3%).
In these clinical studies, the 594 entecavir treated patients who received blinded therapy for more than 52 weeks reported adverse reactions similar in nature and severity to those reported during the first 52 weeks of treatment.
Clinical trials adverse events. Selected clinical adverse events of moderate-severe intensity and considered at least possibly related to treatment occurring during therapy in four clinical studies in which entecavir was compared to lamivudine are presented in Table 2.
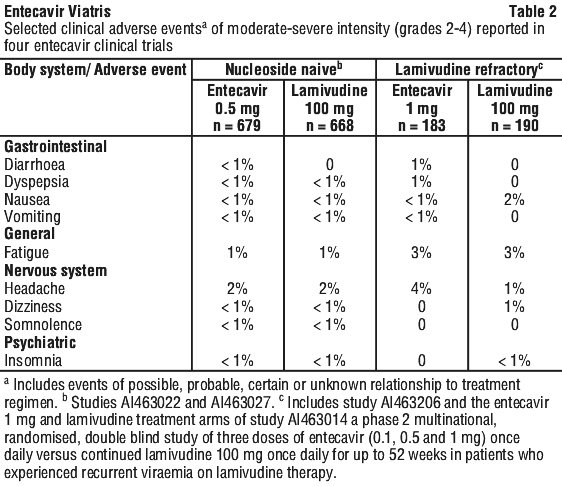
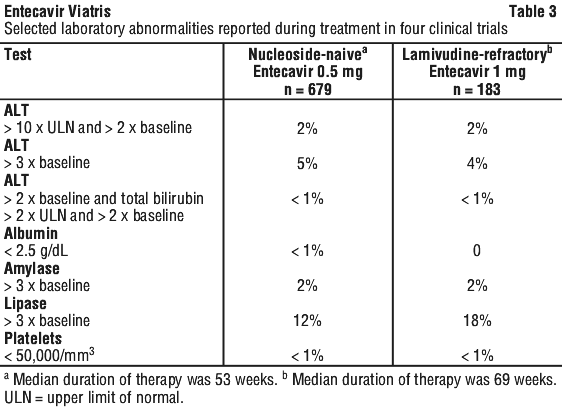
ALT flares after discontinuation of treatment. Acute exacerbations of hepatitis have been reported in patients who have discontinued anti-HBV therapy, including therapy with entecavir (see Section 4.4 Special Warnings and Precautions for Use). The frequency of exacerbations of hepatitis or ALT flare (defined as ALT > 10 x ULN and 2 x the patient's reference level) during off treatment follow-up in clinical studies with entecavir is presented in Table 4.

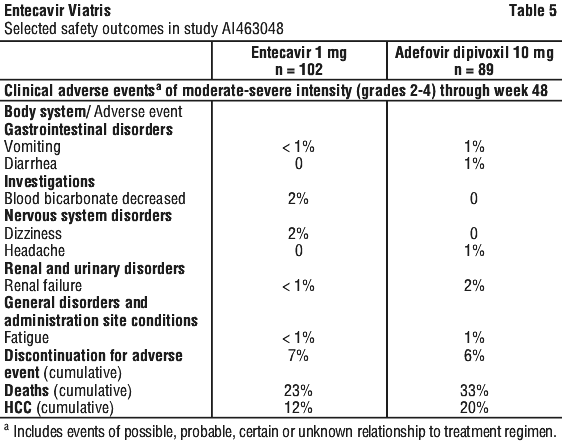
Laboratory test abnormalities reported through week 48 in study AI463048 are listed in Table 6.

Postmarketing experience. The following events have been identified during postapproval use of entecavir. Because reports are voluntary from a population of unknown size, an estimate of frequency cannot be made.
Immune system disorders. Anaphylactoid reaction.
Skin and subcutaneous tissue disorders. Alopecia, rash.
Hepatobiliary disorders. Increased transaminases.
Metabolism and nutrition disorders. Lactic acidosis, often in association with hepatic decompensation, other serious medical conditions or drug exposures.
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
4.9 Overdose
There is limited experience of entecavir overdosage reported in patients. Healthy participants who received single entecavir doses up to 40 mg or multiple doses up to 20 mg/day for up to 14 days had no increase in or unexpected adverse events. If overdose occurs, the patient must be monitored for evidence of toxicity, and standard supportive treatment applied as necessary.
Following a single 1 mg dose of entecavir, a 4 hour haemodialysis session removed approximately 13% of the entecavir dose.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action. Entecavir is a guanosine nucleoside analogue with activity against HBV polymerase. It is phosphorylated to the active triphosphate (TP) form, which has an intracellular half-life of 15 hours. Intracellular TP levels are directly related to extracellular entecavir concentrations, with no significant accumulation beyond initial plateau levels. By competing with the natural substrate deoxyguanosine-TP, entecavir-TP inhibits all 3 functional activities of the viral polymerase: (1) priming of the HBV polymerase, (2) reverse transcription of the negative strand from the pregenomic messenger RNA, and (3) synthesis of the positive strand of HBV DNA. Entecavir-TP Ki value for inhibition of HBV DNA polymerase is 1.2 nanoM. Entecavir-TP is a weak inhibitor of cellular DNA polymerases α, β, δ and ε with Ki values of 18 to 40 microM. It did not inhibit mitochondrial γ polymerase (Ki > 160 microM) and did not affect the mitochondrial DNA content of human hepatoma cells in vitro. Entecavir inhibited HBV DNA synthesis at a concentration of 0.004 microM in human HepG2 cells transfected with wild type HBV. IC50 values for entecavir against lamivudine resistant HBV ranged from 0.029-0.061 microM.
Resistance in vitro. There was reduced susceptibility to entecavir when LVDR substitutions (M204I/V ± L180M) were present. Entecavir inhibited the replication of LVDR HBV at 8-fold higher concentrations than that for the wild type virus in cell based studies. At extracellular concentrations representative of plasma levels achieved with 1 mg dosing, intracellular entecavir-TP levels would be expected to surpass those needed to inhibit the enzyme activity of lamivudine resistant HBV polymerases. Recombinant viruses encoding adefovir resistant substitutions at either rtN236T or rtA181V remained fully susceptible to entecavir.
Lamivudine-resistant strains harbouring rtL180M plus rtM204V in combination with amino acid substitution rtA181C conferred 16- to 122-fold reductions in entecavir phenotypic susceptibility.
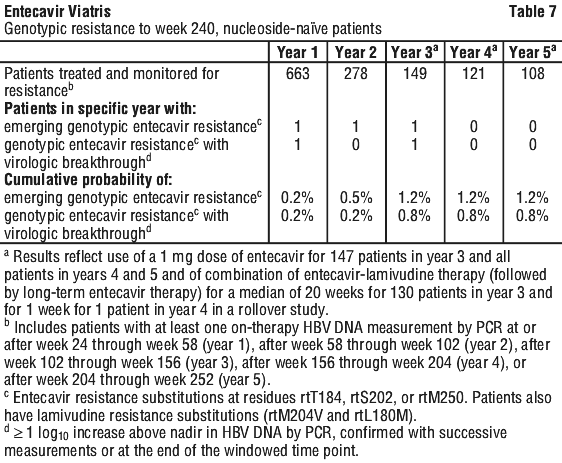
Clinical resistance. Genotyping was performed on paired baseline and on treatment samples from all continuously treated patients with PCR detectable HBV DNA (≥ 300 copies/mL) at week 48, 96, 144, 192 and 240 or at the end of dosing to identify any novel or known resistance substitutions that emerged during entecavir therapy. Virologic breakthrough (≥ 1 log10 increase above nadir in HBV DNA by PCR) due to resistance to entecavir requires the existence of primary lamivudine resistance substitutions (M204I/V ± L180M) along with an additional substitution at residues T184, S202, and/or M250 of the polymerase protein.
Nucleoside naïve studies. Patients in nucleoside naïve studies received 0.5 mg entecavir for up to 96 weeks (see Section 5.1 Pharmacodynamic Properties, Clinical trials). Study participants who failed to achieve the study defined complete response by week 96 were offered continued entecavir treatment in a rollover study. Complete response for HBeAg positive was < 0.7 mEq/mL (approximately 7 x 105 copies/mL) serum HBV DNA and HBeAg loss and, for HBeAg negative, was < 0.7 mEq/mL HBV DNA and ALT normalisation.
By week 96, evidence of emerging amino acid substitution rtS202G with rtM204V and rtL180M was detected in the HBV of 2 subjects and 1 of them experienced virologic rebound (≥ 1 log10 increased above nadir). In addition, emerging amino acid substitution at rtM204I/V and rtL180M, rtL80I or rtV173L, which conferred decreased phenotypic susceptibility to entecavir in the absence of rtT184, rtS202 or rtM250 changes, were detected in the HBV of 3 subjects who experienced virologic rebound.
Results in Table 7 reflect use of a 1 mg dose of entecavir for 147 of 149 patients in year 3 and 121 patients in year 4, 108 patients in year 5 and of combination entecavir plus lamivudine therapy (followed by long-term entecavir monotherapy) for a median of 20 weeks for 130 of 149 patients in year 3 and for 1 week for 1 of 121 patients in year 4 in the rollover study. Through week 240 in nucleoside naïve studies, genotypic evidence of ETVr substitutions at rtT184, rtS202, or rtM250 was identified in 3 patients treated with entecavir, 2 of whom experienced virologic breakthrough (see Table 7). These substitutions were observed only in the presence of LVDr substitutions (rtM204V and rtL180M).
Clinical trials. The safety and efficacy of entecavir were evaluated in three active controlled trials on five continents. These studies included 1633 patients 16 years of age or older with chronic hepatitis B infection (serum HBsAg positive for at least 6 months) accompanied by evidence of viral replication (detectable serum HBV DNA, as measured by the bDNA hybridization or PCR assay). Patients had persistently elevated ALT levels ≥ 1.3 times the upper limit of normal (ULN) and chronic inflammation on liver biopsy compatible with a diagnosis of chronic viral hepatitis. The safety and efficacy of entecavir were also evaluated in an active controlled study of 191 HBV infected patients with decompensated liver disease and in a study of 68 patients coinfected with HBV and HIV.
Nucleoside naïve patients with compensated liver disease. Outcomes at 48 weeks. HBeAg positive. Study AI463022 was a multinational, randomized, double blind study of entecavir tablets 0.5 mg once daily versus lamivudine 100 mg once daily for 52 weeks in 709 (of 715 randomized) nucleoside naïve patients with chronic hepatitis B infection and detectable HBeAg. The mean age of patients was 35 years (range 16 to 78), and 75% were male; 57% were Asian, 40% were Caucasian, and 13% had previously received interferon-α treatment. At baseline, patients had a mean Knodell necroinflammatory score of 7.8, mean serum HBV DNA level as measured by Roche COBAS Amplicor PCR assay of 9.66 log10 copies/mL, and mean serum ALT level of 143 U/L. Response was assessed at week 52 based on test results obtained at the week 48 visit. Ninety six percent of patients had a baseline liver biopsy, paired samples were collected for 89% of patients.
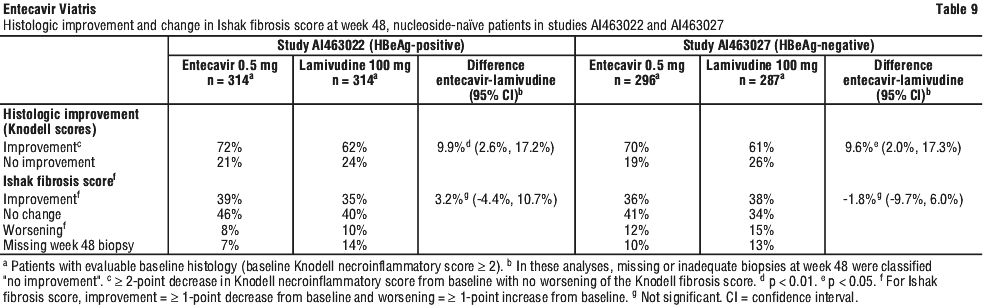
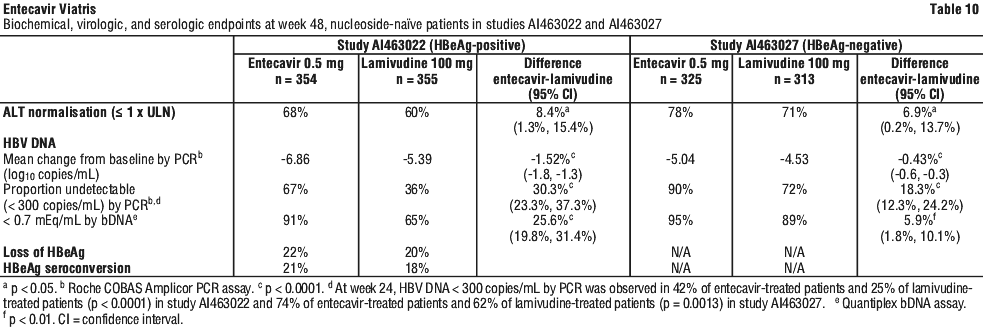
HBeAg negative (anti-HBe positive/HBV DNA positive). Study AI463027 was a multinational, randomized, double blind study of entecavir tablets 0.5 mg once daily versus lamivudine 100 mg once daily for 52 weeks in 638 (of 648 randomized) nucleoside naïve patients with HBeAg negative (HBeAb positive) chronic hepatitis B infection (presumed to have precore or core promoter mutants). The mean age of patients was 44 years (range 18 to 77), and 76% were male. Thirty nine percent were Asian and 58% were Caucasian; 13% had previously received interferon-α treatment. At baseline, patients had a mean Knodell necroinflammatory score of 7.8, mean serum HBV DNA level as measured by Roche COBAS Amplicor PCR assay of 7.58 log10 copies/mL, and mean serum ALT level of 142 U/L. Ninety eight percent of patients had a baseline liver biopsy, and 89% had a biopsy at week 48; paired samples were collected for 88% of patients. Response was assessed at week 52 based on test results obtained at the week 48 visit. In studies AI463022 and AI463027, entecavir was superior to lamivudine on the primary efficacy endpoint of histologic improvement, defined as ≥ 2 point reduction in Knodell necroinflammatory score with no worsening in Knodell fibrosis score at week 48. Histologic improvement and change in Ishak Fibrosis Score are shown in Table 9. Biochemical, virologic, and serologic outcome measures are shown in Table 10.
Covalently closed circular deoxyribonucleic acid (cccDNA) is a stable genomic form of nuclear HBV DNA that serves as a hepatic reservoir of virus, provides the template for HBV transcription, and contributes to viral persistence and relapse. For a subset of patients with paired liver samples in study AI463022, the mean change from baseline in hepatic cccDNA at week 48 was -0.9 log10 copies/human genome equivalents (approximately 8-fold reduction) for entecavir treated patients (n = 159) and -0.7 log10 copies/HGEq (approximately 5-fold reduction) for lamivudine treated patients (n = 146). In study AI463027, the mean change from baseline in hepatic cccDNA at week 48 was -0.5 log10 copies/HGEq (approximately 3-fold reduction) in each treatment group (n = 107 for entecavir treated patients and n = 104 for lamivudine treated patients).
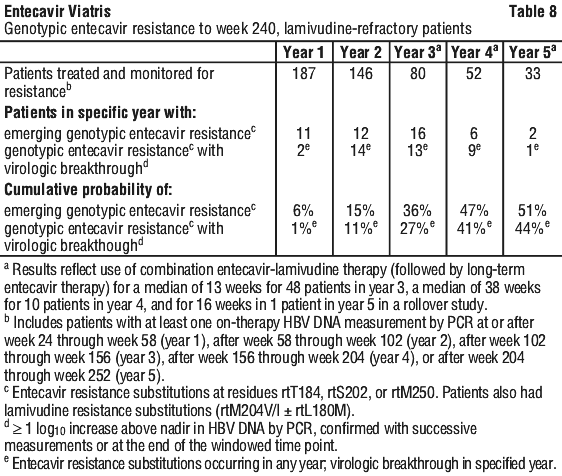
Lamivudine refractory patients. Outcomes at 48 weeks. Study AI463026 was a multinational, randomized, double blind study of entecavir in 286 (of 293 randomized) patients with lamivudine refractory chronic hepatitis B infection. Patients receiving lamivudine at study entry either switched to entecavir 1 mg once daily (with neither a washout nor an overlap period) or continued on lamivudine 100 mg for 52 weeks. The mean age of patients was 39 years (range 16 to 74), and 76% were male; 37% were Asian and 62% were Caucasian. Eighty five percent had LVDR mutations at baseline. Patients had a mean Knodell necroinflammatory score of 6.5, mean serum HBV DNA level as measured by Roche COBAS Amplicor PCR assay of 9.36 log10 copies/mL, and mean serum ALT level of 128 U/L. Response was assessed at week 52 based on test results obtained at the week 48 visit. Ninety eight percent of patients had a baseline liver biopsy, and 88% had a biopsy at week 48; paired samples were collected for 87% of patients.
In study AI463026, entecavir was superior to lamivudine on the coprimary endpoints of histologic improvement (using the Knodell score at week 48) and composite endpoint (proportion of patients with HBV DNA < 0.7 mEq/mL by bDNA assay and ALT < 1.25 x ULN at week 48). These results and change in Ishak Fibrosis Score are shown in Table 11. Table 12 shows biochemical, virologic, and serologic endpoints.
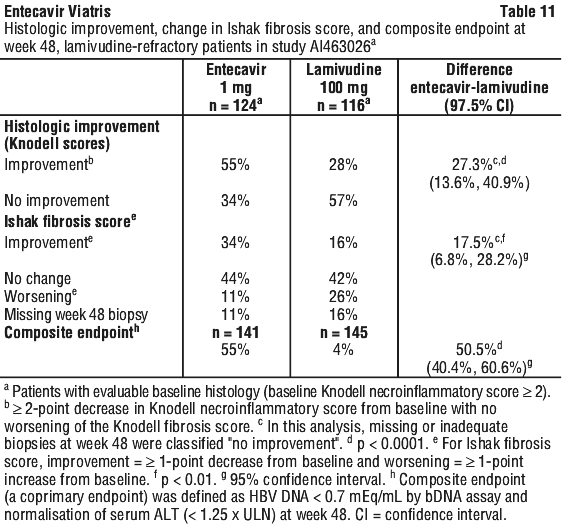
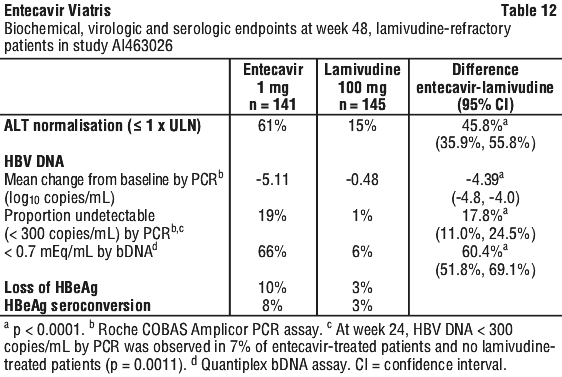
For a subset of patients with paired liver samples in study AI463026, the mean change from baseline in hepatic cccDNA at week 48 was -0.6 log10 copies/HGEq (approximately 4-fold reduction) for entecavir treated patients (n = 74) and 0.0 log10 copies/HGEq for lamivudine treated patients (n = 59).
Health related quality of life (HRQoL) was assessed in study AI463026 using the standardized and validated EQ-5D instrument developed by the EurolQol group. After 48 weeks of therapy, entecavir treated patients experienced significantly less worsening compared to lamivudine treated patients in the dimensions of mobility, self care, and pain/discomfort.
Outcomes beyond 48 weeks. The optimal duration of therapy with entecavir is unknown. According to protocol mandated criteria in the phase 3 clinical trials, participants discontinued entecavir or lamivudine treatment after 52 weeks according to a definition of response based on HBV virologic suppression (< 0.7 mEq/mL by bDNA assay) and either loss of HBeAg in HBeAg positive participants, or ALT < 1.25 x ULN in HBeAg negative participants at week 48. Participants who achieved virologic suppression but did not have serologic response (HBeAg positive) or did not achieve ALT < 1.25 x ULN (HBeAg negative) continued blinded dosing until 96 weeks or until the response criteria were met. These protocol specific participant management guidelines are not intended as guidance for clinical practice.
Nucleoside naïve HBeAg positive. Among nucleoside naïve HBeAg positive participants (study AI463022), 243/354 (69%) entecavir treated participants and 164/355 (46%) lamivudine treated participants continued blinded treatment for up to 96 weeks. Of those continuing blinded treatment in year 2, 180/243 (74%) entecavir treated participants and 60/164 (37%) lamivudine treated participants achieved HBV DNA < 300 copies/mL by PCR at the end of dosing; 193/243 (79%) entecavir treated participants achieved ALT ≤ 1 x ULN compared to 112/164 (68%) lamivudine treated participants. The number of participants with virologic response but not serologic response who achieved a loss of HBeAg at the end of dosing in the second year of blinded treatment was 37/243 (15%) for entecavir and 28/164 (17%) for lamivudine. The proportion who achieved HBeAg seroconversion was 26/243 (11%) for entecavir and 20/164 (12%) for lamivudine.
Post-treatment follow-up: among nucleoside naïve HBeAg positive participants, 111/354 (31%) entecavir treated participants and 93/355 (26%) lamivudine treated participants met the definition of response at end of dosing, discontinued study drugs, and were followed off treatment for up to 24 weeks. In this cohort, 34/111 (31%) entecavir treated patients and 27/93 (29%) lamivudine treated patients had HBV DNA < 300 copies/mL by PCR at the end of follow-up. At the end of follow-up, 78/111 (70%) of the entecavir group and 59/93 (63%) of the lamivudine group recorded ALT ≤ 1 x ULN.
Nucleoside naïve HBeAg negative. Among nucleoside naïve, HBeAg negative participants (study AI463027), 26/325 (8%) entecavir treated participants and 28/313 (9%) lamivudine treated participants continued blinded treatment for up to 96 weeks. In the cohorts continuing treatment, 22/26 entecavir treated and 16/28 lamivudine treated participants had HBV DNA < 300 copies/mL by PCR; 7 entecavir treated and 6 lamivudine treated patients had ALT ≤ 1 x ULN at the end of dosing (up to 96 weeks).
Post-treatment follow-up: among the nucleoside naïve, HBeAg negative participants, 286/325 (88%) treated with entecavir and 253/313 (81%) treated with lamivudine met the definition of response at end of dosing, discontinued study drugs and were followed off treatment for up to 24 weeks. In this cohort, 7/286 (2%) entecavir treated patients and 10/253 (4%) lamivudine treated patients had HBV DNA < 300 copies/mL by PCR at the end of follow-up. At the end of follow-up 129/286 (45%) in the entecavir group and 85/253 (34%) in the lamivudine group recorded ALT ≤ 1 x ULN.
Liver biopsy results. 57 patients from the pivotal nucleoside naïve studies AI463022 (HBeAg positive) and AI463027 (HBeAg negative) who enrolled in a long-term rollover study were evaluated for long-term liver histology outcomes. All 57 patients had both an evaluable baseline and long-term biopsy, with a median duration of entecavir therapy of 280 weeks (approximately 6 years).
55 of 57 (96%) of these patients had histologic improvement (a ≥ 2 point decrease in Knodell necroinflammatory score from baseline with no worsening of the Knodell fibrosis score), and 50 of 57 (88%) had a ≥ 1 point decrease in Ishak fibrosis score. Of the 43 patients with a baseline Ishak fibrosis score of ≥ 2, 25 (58%) had a ≥ 2 point decrease. All (10/10) patients with advanced fibrosis or cirrhosis at baseline (Ishak fibrosis score of 4.5 or 6) had a ≥ 1 point decrease (median decrease from baseline of 1.5 points). At the time of the long-term biopsy, 57 (100%) of patients had HBV DNA < 300 copies/mL and 49 (86%) had serum ALT ≤ 1 x ULN.
Lamivudine refractory. Among lamivudine refractory participants (study AI463026), 77/141 (55%) in the entecavir treated group and 3/145 (2%) in the lamivudine treated group continued blinded treatment for up to 96 weeks. In the entecavir treated cohort, 31/77 (40%) achieved HBV DNA < 300 copies/mL, 62/77 (81%) had ALT ≤ 1 x ULN and 8/77 (10%) demonstrated HBeAg serocoversion at the end of dosing.
Post-treatment follow-up: of the 22/141 (16%) lamivudine refractory patients who met response criteria (HBV DNA < 0.7 mEq/mL on bDNA assay and loss of HBeAg) while receiving entecavir, 5/22 (23%) had HBV DNA < 300 copies/mL by PCR and 12/22 (55%) had ALT ≤ 1 x ULN at the end of follow-up.
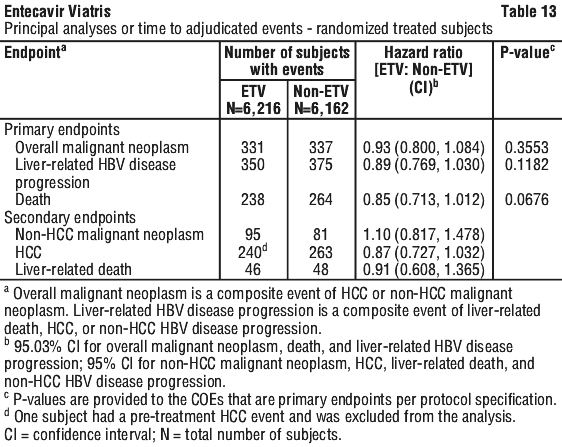
Outcomes of long-term follow-up study. Study AI463080 was a randomized, global, observational, open-label Phase 4 study to assess long-term risks and benefits of entecavir (0.5 mg/day or 1 mg/day) treatment as compared to other standard of care hepatitis B virus nucleoside analogies (nucs) in subjects with chronic HBV (CHB) infection.
A total of 12,378 patients with CHB were treated with ETV (n=6,216) or other standard of care HBV nucleoside (acid) treatment (non-ETV) (n=6,162). The patients were evaluated at baseline and subsequently twice a year (every 6 months) on clinical outcome events (COEs) for up to 10 years during the study. The principal COEs assessed in the study were overall malignant neoplasms, liver-related HBV disease progression, non-HCC malignant neoplasms, HCC, non-HCC HBV disease progression, and deaths, including liver-related deaths. The study data showed that ETV was not significantly associated with an increased risk of malignant neoplasms compared to use of other standard of care HBV nucs, as assessed by either the composite endpoint of overall malignant neoplasms or the individual endpoint of non-HCC malignant neoplasm. The most commonly reported malignancy was HCC followed by gastrointestinal malignancies with colorectal and gastric cancers representing the majority of the observed tumour types within the gastrointestinal system in both ETV and non-ETV groups. The data also showed that long-term ETV use was not associated with a lower occurrence of HBV disease progression or a lower rate of death overall.
There were significant population changes over the long-term follow-up period and more frequent post-randomisation treatment changes in the non-ETV group were noted. The study was underpowered to demonstrate a difference in the non-HCC malignancy rate because of the overall lower than expected background rate.
The principal COE assessment is shown in Table 13:
Liver transplant recipients. The safety and efficacy of entecavir 1 mg once daily were assessed in a single arm, open label study in 65 patients who received a liver transplant for complications of chronic HBV infection and had HBV DNA < 172 IU/mL (approximately 1000 copies/mL) at the time of transplant. The study population was 82% male, 39% Caucasian, and 37% Asian, with a mean age of 49 years; 89% of patients had HBeAg negative disease at the time of transplant. Of the 61 patients who were evaluable for efficacy (received entecavir for at least 1 month), 60 also received hepatitis B immune globulin as part of the post-transplant prophylaxis regimen. At week 72 post-transplant, none of the evaluable patients had HBV recurrence [defined as HBV DNA ≥ 50 IU/mL (approximately 300 copies/mL)] by last observation carried forward analysis.
The frequency and nature of adverse events in this study were consistent with those expected in patients who have received a liver transplant and the known safety profile of entecavir.
5.2 Pharmacokinetic Properties
Absorption. In healthy participants, entecavir was rapidly absorbed with peak plasma concentrations occurring between 0.5 and 1.5 hours. There was a dose proportionate increase in peak plasma concentration (Cmax) and area under the concentration time curve (AUC) values following multiple doses ranging from 0.1 to 1 mg. Steady state was achieved after 6-10 days of once daily dosing with approximately 2-fold accumulation. Cmax and trough plasma concentration (Ctrough) at steady-state were 4.2 and 0.3 nanogram/mL, respectively, for a 0.5 mg dose, and 8.2 and 0.5 nanogram/mL, respectively, for a 1 mg dose. In healthy participants, the bioavailability of the tablet was 100% relative to the oral solution.
Oral administration of entecavir 0.5 mg with a standard high fat meal (945 kcal, 54.6 g fat) or a light meal (379 kcal, 8.2 g fat) resulted in a minimal delay in absorption (1-1.5 hour fed vs. 0.75 hour fasted), a decrease in Cmax of 44-46%, and a decrease in AUC of 18-20%. Therefore, entecavir should be administered on an empty stomach (at least 2 hours before a meal and at least 2 hours after a meal) (see Section 4.2 Dose and Method of Administration).
Distribution. The estimated volume of distribution for entecavir was in excess of total body water, suggesting that it has good penetration into tissues. Protein binding to human serum protein in vitro was approximately 13%.
Metabolism. Entecavir is not a substrate, inhibitor, or inducer of the CYP450 enzyme system. At concentrations approximately 10,000-fold higher than those obtained in humans, entecavir inhibited none of the major human CYP450 enzymes 1A2, 2C9, 2C19, 2D6, 3A4, 2B6, and 2E1. At concentrations approximately 340-fold higher than those observed in humans, entecavir did not induce the human CYP450 enzymes 1A2, 2C9, 2C19, 3A4, 3A5, and 2B6. Following administration of 14C-entecavir in humans and rats, no oxidative or acetylated metabolites and minor amounts of the phase II metabolites glucuronide and sulfate conjugates were observed.
Excretion. After reaching peak levels, entecavir plasma concentrations decreased in a biexponential manner with a terminal elimination half-life of approximately 128-149 hours. The observed drug accumulation index is approximately 2-fold with once daily dosing, suggesting an effective accumulation half-life of about 24 hours.
Entecavir is predominantly eliminated by the kidney with urinary recovery of unchanged drug at steady-state ranging from 62% to 73% of the dose. Renal clearance is independent of dose and ranges between 360 and 471 mL/min suggesting that entecavir undergoes both glomerular filtration and net tubular secretion.
Special populations. Gender/race. The pharmacokinetic profile of entecavir does not vary by gender or race.
Elderly. The pharmacokinetic profile of entecavir does not vary by age.
Renal impairment. The pharmacokinetics of entecavir following a single 1 mg dose were studied in patients (without chronic hepatitis B infection) with selected degrees of renal impairment, including patients whose renal impairment was managed by haemodialysis or continuous ambulatory peritoneal dialysis (CAPD). Results are shown in Table 16.
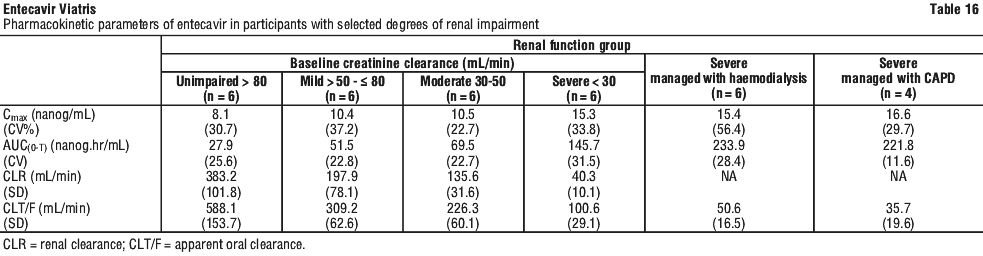
Following a single 1 mg dose of entecavir, haemodialysis removed approximately 13% of the entecavir dose over 4 hours and CAPD removed approximately 0.3% of the dose over 7 days. Entecavir should be administered after haemodialysis.
Hepatic impairment. The pharmacokinetics of entecavir following a single 1 mg dose were studied in patients (without chronic hepatitis B infection) with moderate and severe hepatic impairment. The pharmacokinetics of entecavir were similar between hepatically impaired patients and healthy control participants; therefore, no dosage adjustment of entecavir is recommended for patients with hepatic impairment.
Postliver transplant. Entecavir exposure in HBV infected liver transplant recipients on a stable dose of ciclosporine A (n = 5) or tacrolimus (n = 4) was approximately 2-fold the exposure in healthy participants with normal renal function. Altered renal function contributed to the increase in entecavir exposure in these patients. Before and during entecavir therapy in liver transplant recipients receiving ciclosporine or tacrolimus, renal function should be carefully evaluated (see Section 4.2 Dose and Method of Administration, Renal impairment).
Paediatrics. Pharmacokinetic studies have not been conducted in children.
Drug interactions. Entecavir is not a substrate, inhibitor, or inducer of the CYP450 enzyme system (see Section 5.2 Pharmacokinetic Properties, Metabolism and Excretion). The pharmacokinetics of entecavir are unlikely to be affected by coadministration with agents that are either metabolized by, inhibit, or induce the CYP450 system. Likewise, the pharmacokinetics of known CYP substrates are unlikely to be affected by coadministration of entecavir.
The steady-state pharmacokinetics of entecavir and coadministered drug were not altered in interaction studies of entecavir with each of the following: lamivudine, adefovir dipivoxil, tenofovir disoproxil fumarate (also see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
A pilot study in nine HBV infected liver transplant recipients suggested that concurrent ciclosporine A (n = 5) or tacrolimus (n = 4) therapy did not affect the pharmacokinetics of entecavir (see Section 5.2 Pharmacokinetic Properties, Special populations, Postliver transplant). The effect of entecavir on the pharmacokinetics of ciclosporine A or tacrolimus is unknown.
5.3 Preclinical Safety Data
Genotoxicity. Entecavir was not genotoxic in in vitro assays for bacterial gene mutation, cell transformation and DNA repair, or in an in vivo micronucleus assay for clastogenicity. High concentrations were clastogenic in vitro in human lymphocyte (> 10 microgram/mL) and mouse L5178Y+/- lymphoma cell (> 28 microgram/mL) assays, with evidence that the L5178Y+/- cell response was related to perturbation of cellular deoxyribonucleotide triphosphate pools.
Carcinogenicity. Two year carcinogenicity studies with entecavir were conducted in mice and rats. In mice, entecavir was administered orally at 4 dosage levels representing exposure multiples of 1, 2.4, 10 and 34 times human exposure at the 1 mg dose. The doses in rats achieved exposure multiples relative to the human 1 mg dose of < 0.3, 0.3, 3.9 and 29 times and 0.3, 0.6, 3.6 and 20 times in males and females, respectively. Increases in the incidence of lung tumours were observed in male and female mice at exposures ≥ 2.4 times that in humans. Mechanistic studies suggest the lung tumours are likely to be species specific to mice and probably not relevant to humans. In male rats, entecavir caused pancreatic acinar cell hyperplasia and adenomas at ≥ 3.9 times human exposure. An increase in skin fibromas was seen in female rats at ≥ 3.6 times the exposure in humans at 1 mg/day. The incidence of microglial tumours was increased in rats at and above 0.3 times the exposures in humans at 1 mg/day, reaching statistical significance at 20 times human exposure. Other tumours, which were observed only at exposures ≥ 20 times the exposure in humans at 1 mg/day, included hepatocellular adenomas and/or carcinomas (mice, rates), vascular tumours (mice, rats) salivary duct adenoacanthomas (mice), kidney oncocytoma and malignant mesenchymal tumours (rats), and Zymbal's gland carcinomas (rats; no human counterpart). With the exception of the mouse lung tumours, disruption of cellular deoxyribonucleotide triphosphated (dNTP) pools is likely a significant factor in the carcinogenicity of entecavir, which involves a threshold mechanism. These tumours were generally late appearing and required long-term exposure. Based on animal data, an increased risk of cancer in humans treated with entecavir for an extended period cannot be excluded (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
6 Pharmaceutical Particulars
6.1 List of Excipients
Entecavir film-coated tablets are available for oral administration in strengths of 0.5 mg and 1 mg of entecavir. Entecavir 0.5 mg and 1 mg film-coated tablets contain the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, crospovidone, magnesium stearate, and Opadry white YS-1R-7003 (ARTG PI No. 1625).
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 25°C.
6.5 Nature and Contents of Container
Entecavir Viatris 0.5 mg. Blister pack (PA/Al/PVC/Al) of 30 tablets; bottle (HDPE bottle with PP screw cap) of 30 tablets.
Entecavir Viatris 1 mg. Blister pack (PA/Al/PVC/Al) of 30 tablets; bottle (HDPE bottle with PP screw cap) of 30 tablets.
Some strengths, pack sizes and/or pack types may not be marketed.
Australian register of therapeutic goods (ARTG). AUST R 220088 - Entecavir Viatris entecavir (as monohydrate) 1 mg film coated tablets bottle.
AUST R 220090 - Entecavir Viatris entecavir (as monohydrate) 0.5 mg film coated tablets blister pack.
AUST R 220091 - Entecavir Viatris entecavir (as monohydrate) 1 mg film coated tablets blister pack.
AUST R 220093 - Entecavir Viatris entecavir (as monohydrate) 0.5 mg film coated tablets bottle.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking it to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
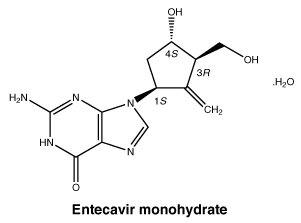
Molecular formula: C12H15N5O3.H2O. Molecular weight: 295.3.
Entecavir monohydrate is a white to off-white crystalline powder. It is slightly soluble in water (2.4 mg/mL), and the pH of the saturated solution in water is 7.9 at 25° ± 0.5°C.
CAS number. 209216-23-9.
7 Medicine Schedule (Poisons Standard)
S4 (Prescription Only Medicine).
Date of First Approval
18 April 2016
Date of Revision
03 November 2022
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.