Epirubicin Accord
Brand Information
| Brand name | Epirubicin Accord |
| Active ingredient | Epirubicin hydrochloride |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Epirubicin Accord.
Summary CMI
EPIRUBICIN ACCORD
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I being given Epirubicin Accord?
Epirubicin Accord contains the active ingredient epirubicin hydrochloride. Epirubicin Accord belongs to a group of medicines known as antineoplastic or cytotoxic agents. Epirubicin Accord is used to treat various types of cancer. It may be used alone or in combination with other medicines.
For more information, see Section 1. Why am I being given Epirubicin Accord? in the full CMI.
2. What should I know before I am given Epirubicin Accord?
Do not use if you have ever had an allergic reaction to any medicine containing Epirubicin, other anthracyclines or anthracenediones or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I am given Epirubicin Accord? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with Epirubicin Accord and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How will I be given Epirubicin Accord?
Epirubicin Accord will be given to you by a doctor or a nurse as a slow injection or a drip (infusion) into a vein. It might also be injected into the bladder.
More instructions can be found in Section 4. How will I be given Epirubicin Accord? in the full CMI.
5. What should I know while being given Epirubicin Accord?
| Things you should do |
|
| Driving or using machines | Be careful driving or operating machinery until you know how Epirubicin Accord affects you. Epirubicin Accord may make some people feel tired or dizzy. |
For more information, see Section 5. What should I know while being given Epirubicin Accord? in the full CMI.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
Some of the serious side effects are:shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue or other parts of the body; rash, itching or hives on the skin; stinging, swelling or pain at the site of injection; flushing of face while the injection is being given; an infection or chills, fever, sore throat, swollen glands, shock; heart problems, fast or irregular heartbeat, shortness of breath; swelling of ankles, feet, legs or hands; bleeding or bruising under the skin; cough, difficulty breathing, chest pain, coughing up blood; swelling, pain, tenderness and redness of the leg.
Tell your doctor immediately even if they occur even if this is several months or years after stopping treatment with Epirubicin Accord.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
EPIRUBICIN ACCORD
Active ingredient(s): Epirubicin hydrochloride
Consumer Medicine Information (CMI)
This leaflet provides important information about using Epirubicin Accord. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using Epirubicin Accord.
Where to find information in this leaflet:
1. Why am I being given Epirubicin Accord?
2. What should I know before I am given Epirubicin Accord?
3. What if I am taking other medicines?
4. How will I be given Epirubicin Accord?
5. What should I know while being given Epirubicin Accord?
6. Are there any side effects?
7. Product details
1. Why am I being given Epirubicin Accord?
Epirubicin Accord contains the active ingredient Epirubicin hydrochloride. Epirubicin Accord belongs to a group of anticancer medicines known as cytotoxic anthracycline antibiotics. You may also hear of these being called chemotherapy medicines. Epirubicin Accord is an anthracycline-type of chemotherapy. Epirubicin Accord works by killing cancer cells and stopping cancer cells from growing and multiplying.
Epirubicin Accord is used to treat various types of cancer. It may be used alone or in combination with other medicines.
Your doctor, however, may prescribe Epirubicin Accord for another purpose.
2. What should I know before I am given Epirubicin Accord?
Warnings
You must not be given Epirubicin Accord if:
- you have an allergy to Epirubicin, other anthracyclines or anthracenediones or any of the ingredients listed at the end of this leaflet.
Do not use the medicine for injection into a vein if you have:
- a low number of red blood cells, white blood cells or platelets in your blood
- sore, red mouth from previous treatment or radiation therapy
- an infection
- severe liver problems
- heart problems or have ever had heart problems
- already received the highest dose allowed for medicines such as mitozantrone, mitomycin C, doxorubicin or daunorubicin
Do not use the medicine for injection into the bladder if you have:
- cancer that has gone into the bladder wall
- kidney or urinary tract infection
- swollen or inflamed bladder
- problems with a catheter (a tube in your bladder)
- blood in the urine
You must tell your doctor if you have or have had:
- heart problems
- liver problems
- kidney problems
- had radiation therapy previously or are having radiation therapy
- been treated previously with medicines to treat cancer
Tell your doctor if you are going to be vaccinated (have an injection to prevent a certain disease)
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Do not use Epirubicin Accord if you are pregnant.
Epirubicin Accord may harm the unborn child.
Tell your doctor if you are planning to have children. Epirubicin Accord may decrease the fertility of men and women.
Use contraception to prevent pregnancy while you or your partner are being treated with Epirubicin Accord.
Epirubicin Accord may cause birth defects if either the male or female is being treated with it. Women being treated must use effective contraceptive methods during treatment and at least 6.5 months after treatment.
Men being treated must use effective contraceptive methods during treatment and for at least 3.5 months after treatment.
Do not use Epirubicin Accord if you are breastfeeding.
You should not breastfeed while taking Epirubicin Accord.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and Epirubicin Accord may interfere with each other. These include:
Medicines used to treat cancer, such as:
- fluorouracil
- cyclophosphamide
- cisplatin
- paclitaxel
- docetaxel
- trastuzumab
- other medicines to treat cancer
Medicines used to treat angina or high blood pressure, such as:
- nifedipine
- verapamil
- diltiazem
- felodipine
- amlodipine
- lercanidipine
- propranolol
A medicine used to treat heartburn or stomach ulcers:
- cimetidine
These medicines and treatments may be affected by Epirubicin Accord or may affect how well it works. You may need different amounts of your medicine, or you may need to have different medicines. Your doctor or pharmacist will advise you.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect Epirubicin Accord.
4. How will I be given Epirubicin Accord?
How much will be given
Epirubicin Accord is usually given as a slow injection or a drip (infusion) into a vein. It might also be injected into the bladder.
Do not drink fluids for 12 hours before treatment if Epirubicin Accord is to be used in the bladder.
Epirubicin Accord may be given alone or in combination with other medicines.
Your doctor will decide the dose of Epirubicin Accord to be given.
When will Epirubicin Accord be given
Epirubicin Accord is usually given every 3 to 4 weeks, in cycles of therapy. However, your doctor may give Epirubicin Accord more or less frequently.
Treatment will not be repeated until your blood counts have returned to acceptable levels and any unwanted effects have been controlled.
Your doctor may change your dose during treatment.
Your doctor will decide how many of these cycles you will need.
If you receive too much Epirubicin Accord
Since Epirubicin Accord is usually given to you in hospital under the supervision of your doctor, it is very unlikely that you will be given too much of the medicine. If you think that you have been given too much Epirubicin Accord.
You should immediately:
- contact your doctor or nurse
- phone the Poisons Information Centre (by calling 13 11 26)
5. What should I know while being given Epirubicin Accord?
Things you must do
Tell your doctor or nurse immediately if the injection stings or hurts while it is being given. The injection may need to be stopped and injected into a different vein.
Be sure to keep all your doctor's appointments. Your doctor will regularly check the function of your heart, liver and kidneys. You will also need to have blood tests.
Use contraception (birth control) to prevent pregnancy while you or your partner are being treated with Epirubicin Accord.
If you become pregnant while you are being given Epirubicin Accord, tell your doctor immediately.
Tell your doctor if you have an infection or fever. Epirubicin Accord lowers your ability to fight infection.
Epirubicin Accord may cause nausea (feeling sick) and vomiting. Tell your doctor if you would like to take medicine to prevent or treat nausea or vomiting.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are being given Epirubicin Accord.
Tell your doctor if you are having or have had radiotherapy.
Tell your doctor if you are having or have had treatment with other anticancer medicines.
Tell your doctor if you have liver problems or kidney problems.
Tell any other doctors, dentists, and pharmacists who treat you that you are being given Epirubicin Accord.
Tell your doctor if you have or have had heart disease or have high blood pressure.
Driving or using machines
Be careful driving or operating machinery or doing jobs that require you to be alert until you know how Epirubicin Accord affects you. Epirubicin Accord may make some people feel tired or dizzy.
Looking after your medicine
The hospital will store Epirubicin Accord under the correct conditions.
Getting rid of any unwanted medicine
Your doctor or pharmacist will dispose of any Epirubicin Accord that may be left over.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Common side effects
| Common side effects | What to do |
| Speak to your doctor if you have any of these common side effects and they worry you |
Serious side effects
| Serious side effects | What to do |
| Call your doctor straight away or go straight to the Emergency Department at your nearest hospital |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed above, such as leukaemia, may also occur in some patients.
After using Epirubicin Accord tell your doctor immediately if you notice any of the following side effects, even if they occur several months or years after stopping treatment with Epirubicin Accord:
- heart problems, fast or irregular heartbeat, shortness of breath
- swelling of ankles, feet, legs or hands, swelling in the stomach
- fever or other signs of infection
Leukaemia may occur after treatment with Epirubicin Accord and other medicines to treat cancer. It is rare.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
Epirubicin Accord does not contain gluten, sucrose, lactose, tartrazine or any other azo dyes.
It is not addictive.
What Epirubicin Accord contains
| Active ingredient (main ingredient) | epirubicin hydrochloride |
| Other ingredients (inactive ingredients) | sodium chloride hydrochloric acid water for injections |
Do not take this medicine if you are allergic to any of these ingredients.
What Epirubicin Accord looks like
Epirubicin Accord is a clear red solution in a glass vial. It is supplied in vials as single packs.
(10 mg/5 mL: AUST R 210084, 20 mg/10 mL: AUST R 210085, 50 mg/25 mL: AUST R 210086, 200 mg/100 mL: AUST R 227997)
Who distributes Epirubicin Accord
Accord Healthcare Pty Ltd
Level 24, 570 Bourke Street
Melbourne, VIC, 3000
Australia
This leaflet was prepared in December 2025.
Brand Information
| Brand name | Epirubicin Accord |
| Active ingredient | Epirubicin hydrochloride |
| Schedule | S4 |
MIMS Revision Date: 01 February 2026
1 Name of Medicine
Epirubicin hydrochloride.
2 Qualitative and Quantitative Composition
1 mL contains 2 mg epirubicin hydrochloride.
1 vial of 5 mL concentrated injection contains 10 mg epirubicin hydrochloride.
1 vial of 10 mL concentrated injection contains 20 mg epirubicin hydrochloride.
1 vial of 25 mL concentrated injection contains 50 mg epirubicin hydrochloride.
1 vial of 100 mL concentrated injection contains 200 mg epirubicin hydrochloride.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Epirubicin hydrochloride concentrated injection is a clear, red solution of epirubicin hydrochloride and sodium chloride in water for injections. Hydrochloric acid is added as necessary to adjust the pH.
4 Clinical Particulars
4.1 Therapeutic Indications
Epirubicin hydrochloride has produced responses in a wide spectrum of neoplastic diseases. Epirubicin hydrochloride injection is indicated for the treatment of breast cancer, gastric cancer, ovarian cancer, small cell lung cancer, lymphoma (non-Hodgkin's lymphoma), advanced/ metastatic soft tissue sarcoma and superficial bladder cancer (Tis, Ta).
In bladder cancer, epirubicin hydrochloride injection is also indicated in the prophylaxis of recurrence after transurethral resection of stage T1 papillary cancers and stage Ta multifocal papillary cancers (grade 2 and 3).
4.2 Dose and Method of Administration
Epirubicin hydrochloride injection is intended for intravenous or intravesical administration only. It must not be administered by the intramuscular, subcutaneous or oral routes.
If epirubicin hydrochloride is administered as a continuous infusion, this should preferably take place via central venous catheter.
Epirubicin Accord is for use in one patient only on one occasion only. Discard any residue.
Dosage. Note. The recommended lifetime cumulative dose limit of epirubicin hydrochloride injection is 900 mg/m2 body surface area.
Intravenous administration. Under conditions of normal recovery from drug-induced toxicity (particularly bone marrow depression and stomatitis), the recommended dosage schedule in adults, as described below, is as a single intravenous injection administered at 21 day intervals.
Standard doses are 75 to 90 mg/m2. Epirubicin hydrochloride injection produces predominantly haematological dose limiting toxicities which are predicted from the known dose response profile of the drug. Based on the patient's haematological status the doctor should determine the choice of dose.
Higher doses, up to 135 mg/m2 as a single agent and 120 mg/m2 in combination, every three (3) to four (4) weeks have been effective in the treatment of breast cancer. In the adjuvant treatment of early breast cancer patients with positive lymph nodes, doses ranging from 100 to 120 mg/m2 every three (3) to four (4) weeks are recommended. Careful monitoring in regard to increased myelosuppression, nausea, vomiting and mucositis are recommended in this high dose setting.
Consideration should be given to the administration of lower starting doses (not exceeding 75 to 90 mg/m2) for heavily pretreated patients, patients with pre-existing bone marrow depression or in the presence of neoplastic bone marrow infiltration. If epirubicin hydrochloride is used in combination with other cytotoxic drugs with potentially overlapping toxicities, the recommended dose/ cycle should be reduced accordingly.
Intravesical administration. For the treatment of papillary transitional cell carcinoma of the bladder, a therapy of eight (8) weekly instillations of 50 mg is recommended.
In the case of local toxicity (chemical cystitis), a dose reduction up to 30 mg is advised. For carcinoma in situ, depending on the individual tolerability of the patient, the dose may be increased up to 80 mg.
For prophylaxis of recurrences after transurethral resection of superficial tumours, four (4) weekly administrations of 50 mg followed by eleven (11) monthly instillations at the same dosage are recommended.
To avoid undue dilution with the urine, the patient should be instructed not to drink any fluid in the 12 hours prior to instillation.
Intravesical administration is not suitable for the treatment of invasive tumours which have penetrated the muscular layer of the bladder wall.
Method of administration. Intravenous administration. Care in the intravenous administration of epirubicin hydrochloride injection will reduce the chance of perivenous infiltration (see Section 4.4 Special Warnings and Precautions for Use, Warnings). It may also decrease the chance of local reactions, e.g. urticaria and erythematous streaking. It is recommended that epirubicin hydrochloride injection be slowly administered into the tubing of a freely running intravenous infusion of Sodium Chloride Injection USP or Glucose Injection 5% USP.
The tubing should be attached to a butterfly needle inserted preferably into a large vein. The rate of administration is dependent on the size of the vein and the dosage. To minimise the risk of thrombosis or perivenous extravasation, the usual infusion times range between 3 and 20 minutes. A direct push injection is not recommended due to the risk of extravasation, which may occur even in the presence of adequate blood return upon needle aspiration. Local erythematous streaking along the vein as well as facial flushing may be indicative of too rapid administration. A burning or stinging sensation may be indicative of perivenous infiltration and the infusion should be immediately terminated and restarted in another vein (see Section 4.4 Special Warnings and Precautions for Use).
The patient's maybe relieved by cooling down the area and keeping it cool for 24 hours. Local infiltration with corticosteroids, with or without the combination of a sodium bicarbonate (8.4%) and local application of dimethyl sulfoxide and cold packs have been used with varying degrees of success. The patient should be monitored closely during the subsequent period of time, as necrosis may occur after several weeks after extravasation occurs.
Intravesical administration. Epirubicin hydrochloride injection, to be instilled using a catheter, should be retained intravesically for one hour. The patient should be instructed to void at the end of this time. During instillation, the pelvis of the patient should be rotated to ensure extensive contact of the solution with the vesical mucosa.
Dose adjustment. Renal impairment. While no specific dose recommendation can be made based on the limited available data in patients with renal impairment, lower starting doses should be considered in patients with severe renal impairment (serum creatinine > 5 mg/dL).
Hepatic impairment. As clinical toxicity may be increased by the presence of impaired liver function, Epirubicin hydrochloride injection dosage must be reduced if hepatic function is impaired according to Table 1.
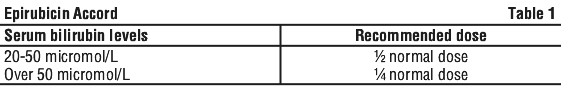
Preparation of solution. See Section 4.4 Special Warnings and Precautions for Use.
Epirubicin hydrochloride can be used in combination with other anti-tumour agents, but it is not recommended that it be mixed with these drugs in the same container (also see Section 6.2 Incompatibilities).
Epirubicin hydrochloride is available as a ready to use solution.
To reduce microbiological hazard, use as soon as practicable after preparation. If storage is necessary, hold at 2°C-8°C for not more than 24 hours.
The product does not contain a preservative. Use once only and discard any residue.
Compatibility. Epirubicin hydrochloride injection is compatible with the following infusion media: 0.9% sodium chloride, 5% glucose, 0.9% sodium chloride with 5% glucose.
Epirubicin hydrochloride injection can be used in combination with other antitumour agents, but it is not recommended that it be mixed with these medicines in the same container.
Pharmaceutical precautions. The following protective recommendations are given due to the toxic nature of this substance.
Personnel should be trained in good technique for handling.
Pregnant staff should be excluded from working with this medicine.
Personnel handling epirubicin hydrochloride should wear protective clothing: goggles, gowns and disposable gloves and masks.
All items used for administration or cleaning, including gloves, should be placed in high risk, waste disposal bags for high temperature incineration.
Spillage or leakage should be treated with dilute sodium hypochlorite (1% available chlorine) solution, preferably by soaking, and then water.
All cleaning materials should be disposed of as indicated previously.
Accidental contact with the eyes or skin should be treated immediately. Copious lavage with water is appropriate treatment for contact with the eyes, whereas water or soap and water, or sodium bicarbonate solution may be used on the skin; medical attention should be sought.
4.3 Contraindications
Hypersensitivity to epirubicin or any other component of the product, other anthracyclines or anthracenediones.
Situations in which patients should not be treated with intravenous epirubicin hydrochloride are:
persisting myelosuppression or severe stomatitis induced by previous drug therapy or radiotherapy;
presence of generalised infections;
marked liver function impairment;
previous history of, or in the presence of, cardiac impairment (severe arrhythmias and cardiomyopathy, previous myocardial infarction);
unstable angina pectoris;
previous treatments with maximum cumulative doses of mitozantrone, mitomycin C or other anthracyclines, such as doxorubicin or daunorubicin;
pregnancy and lactation.
Contraindications for intravesical use are:
invasive tumours that have penetrated the bladder wall;
urinary infections;
inflammation of the bladder;
catheterisation problems;
haematuria.
4.4 Special Warnings and Precautions for Use
Epirubicin hydrochloride should be administered only under the supervision of qualified doctors experienced in cytotoxic therapy.
Patients should recover from acute toxicities (such as stomatitis, neutropenia, thrombocytopenia and generalised infections) of prior cytotoxic treatment before beginning treatment with epirubicin hydrochloride.
While treatment with high doses of epirubicin hydrochloride (e.g. ≥ 90 mg/m2 every 3 to 4 weeks) causes adverse events generally similar to those seen at standard doses (e.g. < 90 mg/m2 every 3 to 4 weeks), the severity of neutropenia and stomatitis/ mucositis may be increased. In particular, treatment with high doses of the drug requires special attention for possible clinical complications due to profound myelosuppression.
Initial treatment with epirubicin hydrochloride requires close observation of the patient and extensive laboratory monitoring including assessment of cardiac function (see Section 4.4 Special Warnings and Precautions for Use, Cardiac function). During each cycle of treatment patients must be carefully and frequently monitored. A blood count, renal and liver function tests should be carried out prior to each epirubicin hydrochloride treatment.
Warnings. Epirubicin hydrochloride injection must be handled with care. If the preparation comes in contact with the skin or mucosae, the appropriate areas should be washed immediately and thoroughly with soap and water or sodium bicarbonate solution.
The rate of administration is dependent on the size of the vein and the dosage. It is important that the dose be administered in not less than three (3) to four (4) minutes. A direct push injection is not recommended due to the risk of extravasation, which may occur even in the presence of adequate blood return upon needle aspiration.
Local erythematous streaking along the vein as well as facial flushing may be indicative of too rapid administration. A burning or stinging sensation may be indicative of perivenous infiltration and the infusion should be immediately terminated and restarted in another vein. Severe local tissue necrosis will occur if there is extravasation during administration. Venous sclerosis may result from infection into a small vessel or from repeated injections into the same vein.
Epirubicin hydrochloride injection must not be given by the intramuscular or subcutaneous route.
Epirubicin hydrochloride is not an antimicrobial agent.
Haematologic toxicity. As with other cytotoxic agents, epirubicin may produce myelosuppression. Haematologic profiles should be assessed before and during each cycle of therapy with epirubicin, including differential white blood cell (WBC) counts. A dose-dependent, reversible leukopaenia and/or granulocytopaenia (neutropaenia) is the predominant manifestation of epirubicin haematologic toxicity and is the most common acute dose-limiting toxicity of this medicine. Leukopaenia and neutropaenia are generally more severe with high-dose schedules, reaching the nadir in most cases between days 10 and 14 after medicine administration; this is usually transient with the WBC/neutrophil counts returning to normal values in most cases by day 21. Thrombocytopaenia and anaemia may also occur.
Clinical consequences of severe myelosuppression include fever, infection, sepsis/ septicaemia, septic shock, haemorrhage, tissue hypoxia or death.
Myelosuppression is more common in patients who have had extensive radiotherapy, bone marrow infiltration by tumour or impaired liver function (when appropriate dosage reduction has not been adopted) (see Section 4.2 Dose and Method of Administration, Dose adjustment, Other special populations).
Secondary leukaemia. Secondary leukaemia, with or without a pre-leukaemic phase, has been reported in patients treated with anthracyclines including epirubicin. Secondary leukaemia is more common when such medicines are given in combination with DNA-damaging antineoplastic agents, when patients have been heavily pre-treated with cytotoxic drugs, or when doses of the anthracyclines have been escalated. These leukaemias can have a 1 to 3-year latency period.
Cardiac function. Cardiotoxicity is a risk of anthracycline treatment that may be manifested by early (i.e. acute) or late (i.e. delayed) events. The cardiac abnormalities caused by treatment can be separated into 2 categories:
(i) ECG alterations and,
(ii) Congestive heart failure (CHF).
Early (i.e. acute) events. Early cardiotoxicity of epirubicin consists mainly of sinus tachycardia and/or electrocardiogram (ECG) abnormalities such as non-specific ST-T wave changes. ECG changes following epirubicin hydrochloride treatment occur in about 10% of patients. Tachyarrhythmias, including premature ventricular contractions, ventricular tachycardia, and bradycardia, as well as atrioventricular and bundle branch block have also been reported. These effects do not usually predict subsequent development of delayed cardiotoxicity, are rarely of clinical importance, and are generally not a consideration for the discontinuation of epirubicin treatment.
Late (i.e. delayed) events. Delayed cardiotoxicity usually develops late in the course of therapy with epirubicin hydrochloride or within two to three months post treatment termination, but later events several months to years after completion of treatment have also been reported. Cardiomyopathy induced by anthracyclines is associated with persistent QRS voltage reduction, prolongation beyond normal limits of the systolic time interval (PEP/LVET) and a reduction of the ejection fraction and/or signs and symptoms of congestive heart failure (CHF), e.g. dyspnoea, pulmonary oedema, dependent oedema, cardiomegaly and hepatomegaly, oliguria, ascites, pleural effusion and gallop rhythm. Life threatening CHF is the most severe form of anthracycline induced cardiomyopathy and represents the cumulative dose limiting toxicity of the drug. Pericardial effusion has also been described.
The risk of developing CHF increases rapidly with increasing total cumulative doses of epirubicin in excess of 900 mg/m2; this cumulative dose should only be exceeded with extreme caution.
Cardiac function should be assessed before patients undergo treatment with epirubicin and must be monitored throughout therapy to minimise the risk of incurring severe cardiac impairment. The risk may be decreased through regular monitoring of left ventricular ejection fraction (LVEF) during the course of treatment with prompt discontinuation of epirubicin at the first sign of impaired function. The appropriate quantitative method for repeated assessment of cardiac function (evaluation of LVEF) includes multi-gated radionuclide angiography (MUGA) or echocardiography (ECHO). A baseline cardiac evaluation with an ECG and either a MUGA scan or an ECHO is recommended, especially in patients with risk factors for increased cardiotoxicity. Repeated MUGA or ECHO determinations of LVEF should be performed, particularly with higher, cumulative anthracycline doses. The technique used for assessment should be consistent throughout follow-up.
Risk factors for cardiac toxicity include active or dormant cardiovascular disease, concomitant or previous radiation of the mediastinal-pericardial area, hypertensive cardiomyopathy, previous therapy with other anthracyclines or anthracenediones, concomitant use of other drugs with the ability to suppress cardiac contractility or cardiotoxic agents (e.g. trastuzumab, high dose cyclophosphamide or 5-fluorouracil). Anthracyclines including epirubicin should not be administered in combination with other cardiotoxic agents unless the patient's cardiac function is closely monitored (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). Patients receiving anthracyclines after stopping treatment with other cardiotoxic agents, especially those with long half-lives such as trastuzumab, may also be at an increased risk of developing cardiotoxicity. The half-life of trastuzumab is variable. Trastuzumab may persist in the circulation for up to 7 months. Therefore, physicians should avoid anthracycline-based therapy for up to 7 months after stopping trastuzumab when possible. If anthracyclines are used before this time, careful monitoring of cardiac function is recommended.
Cardiac function monitoring must be particularly strict in patients receiving high cumulative doses and in those with risk factors. However, cardiotoxicity with epirubicin may occur at lower cumulative doses whether or not cardiac risk factors are present.
There have been sporadic reports of foetal/neonatal cardiotoxic events including fetal death following in utero exposure to epirubicin (see Section 4.6 Fertility, Pregnancy and Lactation).
It is probable that the toxicity of epirubicin and other anthracyclines or anthracenediones is additive.
Gastrointestinal. Epirubicin is emetogenic. Nausea and vomiting may be prevented or alleviated by the administration of appropriate antiemetic therapy.
Mucositis/ stomatitis occurs frequently and generally appears early after drug administration, most commonly developing 5 to 10 days after treatment. It is painful and typically begins as a burning sensation in the mouth and pharynx. The mucositis may involve the vagina, rectum and oesophagus, and, if severe, may progress over a few days to mucosal ulcerations with a risk of secondary infection. Most patients recover from this adverse event by the third week of therapy.
Use in hepatic impairment. The major route of elimination of epirubicin is the hepatobiliary system. Serum total bilirubin and AST levels should be evaluated before and during treatment with epirubicin hydrochloride. Patients with elevated bilirubin or AST may experience slower clearance of medicine with an increase in overall toxicity. Lower doses are recommended in these patients (see Section 4.2 Dose and Method of Administration). Patients with severe hepatic impairment should not receive epirubicin hydrochloride (see Section 4.3 Contraindications).
Use in renal impairment. Moderate renal impairment does not appear to require a dose reduction in view of the limited amount of epirubicin hydrochloride excreted by this route. However, serum creatinine should be assessed before and during therapy as dosage adjustment is necessary in patients with serum creatinine > 5 mg/dL (see Section 4.2 Dose and Method of Administration).
Effects at site of injection. Phlebosclerosis may result from an injection into a small vessel or from repeated injections into the same vein. Following the recommended administration procedures may minimise the risk of phlebitis/ thrombophlebitis at the injection site (see Section 4.2 Dose and Method of Administration, Intravenous administration).
Extravasation. Extravasation of epirubicin during intravenous injection may produce local pain, severe tissue lesions (vesication, severe cellulitis) and necrosis. The recommended administration procedures should be followed (see Section 4.2 Dose and Method of Administration, Intravenous administration). Should signs or symptoms of extravasation occur during intravenous administration of epirubicin, the drug infusion should be immediately stopped.
Tumour-lysis syndrome. Epirubicin may induce hyperuricaemia because of the extensive purine catabolism that accompanies rapid drug-induced lysis of neoplastic cells (tumour-lysis syndrome). Blood uric acid levels, potassium, calcium phosphate and creatinine should be evaluated after initial treatment. Hydration, urine alkalinisation and prophylaxis with allopurinol to prevent hyperuricaemia may minimise potential complications of tumour-lysis syndrome.
Immunosuppressant effects/ increased susceptibility to infections. Administration of live or live-attenuated vaccines in patients immunocompromised by chemotherapeutic agents, including epirubicin, may result in serious or fatal infections. Vaccination with a live vaccine should be avoided in patients receiving epirubicin. Killed or inactivated vaccines may be administered; however, the response to such vaccines may be diminished.
Embryo-fetal toxicity. Epirubicin can cause genotoxicity. An effective method of contraception is required for both male and female patients during and for a period after treatment with epirubicin (see Section 4.6 Fertility, Pregnancy and Lactation). Patients desiring to have children after completion of therapy should be advised to obtain genetic counselling if appropriate and available.
Other. As with other cytotoxic agents, thrombophlebitis and thromboembolic phenomena, including pulmonary embolism (in some cases fatal), have been coincidently reported with the use of epirubicin hydrochloride.
Epirubicin hydrochloride may enhance radiation induced toxicity such as skin reactions and mucositis and may potentiate the toxicity of other anticancer therapies. This has to be taken into account particularly when using the drug in high doses and the availability of supportive care and facilities has to be considered before initiating high dose intensive regimens.
Epirubicin may impart a red colour to the urine for one (1) to two (2) days after administration. Patients should be advised that such an event is not a cause for alarm.
Intravesical route. Administration of epirubicin may produce symptoms of chemical cystitis (such as dysuria, polyuria, nocturia, stranguria, haematuria, bladder discomfort, necrosis of the bladder wall) and bladder constriction. Special attention is required for catheterisation problems (e.g. uretheral obstruction due to massive intravesical tumours).
Use in the elderly. No data available.
Paediatric use. No data available.
Effects on laboratory tests. No data available.
4.5 Interactions with Other Medicines and Other Forms of Interactions
Epirubicin hydrochloride is mainly used in combination with other cytotoxic drugs and additive toxicity may occur especially with regard to bone marrow/ haematological and gastrointestinal effects. In addition, the concomitant use of epirubicin hydrochloride with other antitumour drugs which have been reported as potentially cardiotoxic (e.g. fluorouracil, cyclophosphamide, cisplatin, taxanes, trastuzumab), as well as the concomitant use of other cardioactive compounds (e.g. calcium channel blockers), require a close monitoring of cardiac function throughout treatment.
Propranolol: concurrent administration of epirubicin hydrochloride and propranolol may result in an additive cardiotoxic effect.
Cimetidine increased the AUC of epirubicin hydrochloride by 50% and should be stopped during treatment with epirubicin hydrochloride.
When given prior to epirubicin, paclitaxel can cause increased plasma concentrations of unchanged epirubicin. Co-administration of paclitaxel or docetaxel did not affect the pharmacokinetics of epirubicin when epirubicin was administered prior to taxane.
Concurrent mediastinal radiotherapy and epirubicin hydrochloride may be associated with enhanced myocardial toxicity of epirubicin hydrochloride.
Epirubicin hydrochloride is extensively metabolised by the liver. Changes in hepatic function induced by concomitant therapies may affect epirubicin hydrochloride metabolism, pharmacokinetics, therapeutic efficacy and/or toxicity.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. Studies in animals have shown reproductive toxicity. In fertility studies in rats, males were given epirubicin daily for 9 weeks and mated with females that were given epirubicin daily for 2 weeks prior to mating and through Day 7 of gestation. When 0.3 mg/kg/day (0.01-times the maximum recommended clinical dose based on body surface area, BSA) was administered to both sexes, no pregnancies resulted. No effects on mating behaviour or fertility were observed at 0.1 mg/kg/day, but male rats had atrophy of the testes and epididymis, and reduced spermatogenesis. Multiple daily doses of epirubicin to rabbits and dogs also caused atrophy of male reproductive organs. Single intravenous doses of epirubicin 20.5 and 12 mg/kg caused testicular atrophy in mice and rats, respectively. A single-dose of 16.7 mg/kg epirubicin caused uterine atrophy in rats.
In women, epirubicin hydrochloride may cause amenorrhea. After termination of therapy, ovulation and menstruation may be expected to return in a few months, often accompanied by normal fertility. Premature menopause may also occur.
In male patients, oligospermia or azoospermia may be permanent, although fertility may return several years after ceasing therapy. Given the mutagenic potential of epirubicin hydrochloride, the drug could induce chromosomal damage in human spermatozoa; therefore, males undergoing epirubicin hydrochloride treatment should be advised to use effective contraceptive methods during treatment and for at least 3.5 months after the last dose.
Based on animal studies, male and female fertility may be compromised. It is recommended to discuss fertility preservation with men and women prior to treatment.
Use in pregnancy. (Category D)
Women of child-bearing potential should be advised to avoid becoming pregnant during treatment and to use effective contraceptive methods during treatment and for at least 6.5 months after last dose.
There is no specific information available at present concerning the use of epirubicin hydrochloride in human pregnancy. However, as it has been shown to be embryotoxic and fetotoxic in animals, it should not be used in patients who are pregnant or are likely to become pregnant.
Administration of 0.8 mg/kg/day intravenously (0.04-times the maximum recommended clinical dose based on body surface area) of epirubicin to rats during days 5 to 15 of gestation was embryotoxic (increased resorptions and post-implantation loss) and caused fetal growth retardation (decreased body weight), but was not teratogenic up to this dose. Administration of 2 mg/kg/day intravenously (0.08 times the maximum recommended clinical dose based on body surface area) of epirubicin to rats on days 9 and 10 of gestation was embryotoxic (increased late resorptions, post-implantation losses, and dead fetuses; and decreased live fetuses), retarded fetal growth (decreased body weight), and caused decreased placental weight. This dose was also teratogenic, causing numerous external (anal atresia, misshapen tail, abnormal genital tubercle), visceral (primarily gastrointestinal, urinary, and cardiovascular systems), and skeletal (deformed long bones and girdles, rib abnormalities, irregular spinal ossification) malformations. Administration of intravenous epirubicin to rabbits at doses up to 0.2 mg/kg/day (0.02 times the maximum recommended clinical dose based on body surface area) during days 6 to 18 of gestation was not embryotoxic or teratogenic, but a maternally toxic dose of 0.32 mg/kg/day increased abortions and delayed ossification. Administration of a maternally toxic intravenous dose of 1 mg/kg/day epirubicin (0.08 times the maximum recommended clinical dose based on body surface area) to rabbits on days 10 to 12 of gestation induced abortion, but no other signs of embryofetal toxicity or teratogenicity were observed. When doses up to 0.5 mg/kg/day epirubicin (0.02 times the maximum recommended clinical dose based on body surface area) were administered to rat dams from Day 17 of gestation to Day 21 after delivery, no permanent changes were observed in the development, functional activity, behaviour, or reproductive performance of the offspring.
If epirubicin is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus. There have been sporadic reports of foetal and/or neonatal transient ventricular hypokinesia, transient elevation of cardiac enzymes, and of fetal death from suspected anthracycline-induced cardiotoxicity following in utero exposure to epirubicin in 2nd and/or 3rd trimesters (see Section 4.4 Special Warnings and Precautions for Use). Monitor the foetus and/or neonate for cardiotoxicity and perform testing consistent with community standards of care.
Use in lactation. In a peri- and post-natal study, epirubicin was present in milk of rats treated with 0.5 mg/kg/day epirubicin (0.02 times the maximum recommended clinical dose based on body surface area).
It is likely that epirubicin hydrochloride is excreted in breast milk in humans, therefore, it is not recommended for nursing mothers unless the expected benefit outweighs any potential risk. Because of the potential for serious adverse reactions in nursing infants from epirubicin, lactating women should be advised not to breastfeed during treatment with epirubicin and for at least 7 days after last dose.
4.7 Effects on Ability to Drive and Use Machines
The effects of this medicine on a person's ability to drive and use machines were not assessed as part of its registration.
4.8 Adverse Effects (Undesirable Effects)
Dose limiting toxicities are myelosuppression and cardiotoxicity (see Section 4.4 Special Warnings and Precautions for Use).
Adverse effects observed are:
More common reactions > 5%. Blood and lymphatic system disorders. Myelosuppression, leukopenia, neutropenia, febrile neutropenia, thrombocytopenia, mild anaemia, secondary infection.
Cardiac disorders. Transient ECG changes, including low QRS voltage, tachycardia, arrhythmias, T wave flattening, ST depression and T inversion.
Gastrointestinal disorders. Nausea, vomiting, diarrhoea, mucositis (erythema, erosions/ ulcerations, bleeding). Mucositis may appear 5-10 days after the start of treatment and usually involves stomatitis with areas of painful erosions, mainly along the sides of the tongue and on the sublingual mucosa.
Skin and subcutaneous tissue disorders. Alopecia, including the interruption of beard growth, usually reversible, occurs in 60 - 90% of treated cases.
General disorders and administration site conditions. Erythematous streaking along the infused vein.
Metabolism and nutrition disorders. Dehydration.
Less common reactions < 5%. Blood and lymphatic system disorders. Severe thrombocytopenia, anaemia, severe myelosuppression, pancytopenia, sepsis, septicaemia, septic shock, tissue hypoxia, haemorrhage and death.
Cardiac disorders. Cardiomyopathy, congestive heart failure, cardiomegaly, atrioventricular and bundle branch block, tachyarrhythmias (premature ventricular contractions, ventricular tachycardia, bradycardia).
Gastrointestinal disorders. Oesophagitis, bleeding, hyperpigmentation of oral mucosa and abdominal pain or burning sensation.
Skin and subcutaneous tissue disorders. Local toxicity, rash/ itch, transient urticaria, erythema, flushes, skin and nail hyperpigmentation, photosensitivity and hypersensitivity of irradiated skin.
General disorders and administration site conditions. Chills, fever, malaise/asthenia, vesication, local pain, severe cellulitis and skin necrosis following perivenous drug extravasation.
Eye disorders. Conjunctivitis, keratitis.
Immune system disorders. Anaphylaxis.
Investigations. Asymptomatic drops in left ventricular ejection fraction, changes in transaminase levels.
Nervous system disorders. Weakness, dizziness, confusion, depression, paraesthesia.
Metabolism and nutrition disorders. Hyperuricaemia, anorexia.
Neoplasms benign and malignant. Acute lymphocytic leukaemia, acute myelogenous leukaemia.
Reproductive system disorders. Amenorrhoea, azoospermia.
Vascular disorders. Hot flushes, shock, thromboembolism, arterial embolism, thrombophlebitis, phlebitis, venous sclerosis.
Post-marketing experience. Infections and infestations. Pneumonia.
Renal and urinary disorders. Red coloration of urine for 1 to 2 days after administration.
Respiratory, thoracic and mediastinal disorders. Pulmonary embolism.
Injury, poisoning and procedural complications. Chemical cystitis, sometimes haemorrhagic, and bladder constriction (following intravesical administration). Dose reduction (40%) may be necessary in these cases.
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
4.9 Overdose
A 36-year-old man with non-Hodgkin's lymphoma received a daily 95 mg/m2 dose of epirubicin injection for 5 consecutive days. Five days later, he developed bone marrow aplasia, grade 4 mucositis and gastrointestinal bleeding. No signs of acute cardiac toxicity were observed. He was treated with antibiotics, colony-stimulating factors and antifungal agents and recovered completely. A 63-year-old woman with breast cancer and liver metastasis received a single 320 mg/m2 dose of epirubicin, which resulted in hyperthermia, multiple organ failure (respiratory and renal), lactic acidosis, increased lactate dehydrogenase and anuria, and death within 24 hours of administration.
Additional instances of administration of doses higher than recommended have been reported at doses ranging from 150 to 250 mg/m2. The observed adverse events in these patients were qualitatively similar to known toxicities of epirubicin. Most of the patients recovered with appropriate supportive care.
Symptoms. Very high single doses of epirubicin hydrochloride may be expected to cause acute myocardial degeneration within 24 hours, and severe myelosuppression (mainly leukopenia and thrombocytopenia) within 10 to 14 days and also gastrointestinal toxic effects (mainly mucositis).
Treatment. If an overdose occurs, supportive treatment (including antibiotic therapy, blood and platelet transfusions, colony-stimulating factors and intensive care as needed) should be provided until the recovery of toxicities. Delayed cardiac failure may occur up to six (6) months after the overdose. Patients should be observed carefully and should, if signs of cardiac failure arise, be treated along conventional lines.
Epirubicin cannot be removed by dialysis.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action. The mechanism of action of epirubicin hydrochloride has not been fully elucidated but is probably related to its ability to bind deoxyribonucleic acid (DNA). Cell culture studies have shown cell penetration, localisation in the nucleus and inhibition of nucleic acid synthesis and mitosis. Epirubicin hydrochloride has proved to be active on the following experimental tumours: L 1210 ascites and P388 leukaemias, sarcoma SA 180 (solid and ascitic forms), melanoma B 16, mammary carcinoma, Lewis lung carcinoma and colon carcinoma 38.
The specificity of epirubicin hydrochloride toxicity appears to be related primarily to proliferative activity of normal tissue. Thus, bone marrow, gastrointestinal tract, lymphoid organs and the gonads are the main normal tissues damaged. Degenerative or functional alterations in liver and kidneys were also seen in animals dosed with epirubicin hydrochloride.
Toxicity studies in animals have indicated that on a weight (mg per mg) basis epirubicin hydrochloride has a better therapeutic index and less systemic and cardiac toxicity than doxorubicin.
Epirubicin hydrochloride is immunosuppressive in animals. Although there are no clinical data on the immunosuppressive effects of epirubicin hydrochloride, effects similar to those seen with doxorubicin may be expected.
Clinical trials. Early breast cancer. Data from two multicentre, randomised phase III studies support the use of epirubicin hydrochloride 100 to 120 mg/m2 for the adjuvant treatment of patients with axillary node positive breast cancer and no evidence of distant metastatic disease (stage II or III). In one study, an intensive cyclophosphamide/ epirubicin/ fluorouracil (CEF-120) regimen (epirubicin given in a dose of 60 mg/m2 on days 1 and 8) was compared with a conventional cyclophosphamide/ methotrexate/ fluorouracil (CMF) regimen. A total of 716 patients were randomised, 356 to CEF and 360 to CMF. Both disease free survival and overall survival were significantly prolonged in the CEF arm at five years. Disease free survival was 62% for CEF versus 53% for CMF (p = 0.01) and overall survival was 77% for CEF versus 70% for CMF (p = 0.043).
In the second study, 301 patients were randomised to receive tamoxifen 20 mg/day alone for four (4) years and 303 patients were randomised to receive tamoxifen for four (4) years in combination with epirubicin 50 mg/m2 on days 1 and 8 every four (4) weeks for six (6) cycles. Although there was no significant difference between the two arms with regard to disease free survival and overall survival, there was a trend in favour of the combined use of epirubicin and tamoxifen. Disease free survival at two years was 85.1% versus 77.9%, and at five years was 70.4% versus 59.5% (p = 0.07). Overall survival at two (2) years was 93% versus 92% and five (5) years was 78.8% versus 72.9%.
Advanced breast cancer. Data from four (4) open label, multicentre, phase III studies support the use of epirubicin hydrochloride for the treatment of patients with locally advanced or metastatic breast cancer. In study 1, an intensified cyclophosphamide/ epirubicin/ fluorouracil (CEF-100) regimen (epirubicin given in a dose of 50 mg/m2 on days 1 and 8) was compared with a conventional CMF regimen (n = 461). Studies 2 and 3 compared cyclophosphamide/ epirubicin/ fluorouracil regimens where only the dose of epirubicin varied. In both of these, epirubicin was given in a dose of 50 mg/m2 on day 1 and compared with either 100 mg/m2 on day 1 (n = 456) or 50 mg/m2 on days 1 and 8 (n = 164). High dose epirubicin (135 mg/m2) was compared to conventional dose epirubicin (75 mg/m2) in study 4 (n = 151).
The efficacy endpoints included response rate (RR), duration of response (DR), time to tumour progression (TTP), time to treatment failure (TTF), and overall survival (OS). In study 1, the CEF-100 regimen produced a significantly higher RR, a significantly longer TTP and a significantly longer TTF than the CMF regimen. In studies 2, 3 and 4, the higher dose epirubicin containing regimens produced a significantly greater RR than the lower dose epirubicin containing regimens. DR and TTF were also significantly longer in study 3 and TPP was significantly longer in study 4 for the higher dose epirubicin regimens.
5.2 Pharmacokinetic Properties
There is evidence for a dose response and dose toxicity relationship for epirubicin in breast cancer and, to a lesser extent, for lymphoma. This relationship is steeper and therefore more evident for doses of epirubicin above 90 mg/m2. Current data indicate that an increase in dose (for dose intensity) produces greater response rates.
Absorption. When epirubicin hydrochloride is administered intravesically, the systemic absorption is minimal.
Distribution. As with doxorubicin, epirubicin hydrochloride may not be expected to cross the blood brain barrier.
Metabolism. In patients with normal hepatic and renal function, plasma levels after intravenous injection of 75 to 90 mg/m2 of the drug follow a triexponential decreasing pattern with a very fast first phase and a slow terminal phase with a mean half-life of about 40 hours. Plasma levels of the drug's main metabolite, the 13-OH derivative, are constantly somewhat lower and virtually parallel to those of the unchanged medicine.
Excretion. Epirubicin hydrochloride is eliminated mainly through the liver; high plasma clearance values (0.9 L/minute), indicate that the slow elimination of epirubicin is due to extensive tissue distribution. Urinary excretion accounts for approximately 11% of the administered dose in 48 hours. However, like doxorubicin, biliary excretion is likely to be the major excretion route. Impairment of liver function delays plasma clearance.
5.3 Preclinical Safety Data
Genotoxicity. Epirubicin was mutagenic in vitro to bacteria (Ames test) either in the presence or absence of metabolic activation and to mammalian cells (HGPRT assay in V79 Chinese hamster lung fibroblasts) in the absence but not in the presence of metabolic activation. Epirubicin was clastogenic in vitro (chromosome aberration in human lymphocytes) both in the presence and absence of metabolic activation and was also clastogenic in vivo (chromosome aberration in mouse bone marrow).
Carcinogenicity. Epirubicin hydrochloride is carcinogenic in animals. Conventional long-term animal studies to evaluate the carcinogenic potential of epirubicin have not been conducted, but intravenous administration of a single 3.6 mg/kg epirubicin dose (0.16 times the maximum recommended clinical dose based on body surface area) to female rats approximately doubled the incidence of mammary tumours (primarily fibroadenomas) observed at 1 year. Administration of 0.5 mg/kg epirubicin intravenously (0.02 times the maximum recommended clinical dose based on body surface area) to rats every 3 weeks for ten doses increased the incidence of subcutaneous fibromas in males over an 18-month observation period. In addition, subcutaneous administration of 0.75 or 1.0 mg/kg/day to newborn rats for 4 days on both the first and tenth day after birth for a total of eight doses increased the incidence of animals with tumours compared to controls during a 24-month observation period.
6 Pharmaceutical Particulars
6.1 List of Excipients
Sodium chloride, hydrochloric acid, water for injections.
6.2 Incompatibilities
If epirubicin hydrochloride is used in combination with other anti-tumour agents, it should not be mixed with these drugs in the same container.
Epirubicin hydrochloride should not be mixed with other drugs. Contact with any solution of an alkaline pH should be avoided, as it will result in hydrolysis of epirubicin.
Epirubicin hydrochloride should not be mixed with heparin as these drugs are incompatible.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store at 2°C to 8°C. Refrigerate, do not freeze. Protect from light.
6.5 Nature and Contents of Container
Epirubicin Accord is available in four strengths containing the active ingredient epirubicin hydrochloride as follows: 10 mg/5 mL, 20 mg/10 mL, 50 mg/25 mL and 200 mg/100 mL in type I clear glass vials in packs of 1.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Structurally, epirubicin hydrochloride differs from doxorubicin hydrochloride only in the orientation of the hydroxyl group at the 4 position on the aminoglycoside ring. Epirubicin hydrochloride is a red orange, almost odourless, hygroscopic powder, sparingly soluble in water and dilute alcohol.
Chemical name: (8S, 10S)-10-[(3-amino-2,3,6-trihydroxy-alpha-L-arabino -hexopyranosyl) oxy]-6,8,11-trihydroxy-8 (hydroxyacetyl)-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione hydrochloride).
Molecular formula: C27H29NO11.HCl.
Molecular weight: 579.99.
Chemical structure.
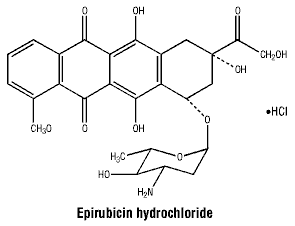
7 Medicine Schedule (Poisons Standard)
S4 - Prescription Only Medicine.
Date of First Approval
09 October 2014
Date of Revision
22 December 2025
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.