Kanuma
Brand Information
| Brand name | Kanuma |
| Active ingredient | Sebelipase alfa |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Kanuma.
Summary CMI
KANUMA®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I using Kanuma?
Kanuma contains the active ingredient sebelipase alfa rce. Sebelipase alfa rce is similar to the naturally occurring enzyme lysosomal acid lipase (LAL), which the body uses to breakdown fats. Kanuma is used to treat patients of all ages with lysosomal acid lipase deficiency (LAL-D).
For more information, see Section 1. Why am I using Kanuma? in the full CMI.
2. What should I know before I use Kanuma?
Do not use if you or your child has had a life-threatening allergic reaction to Kanuma, or to egg, or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use Kanuma? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with Kanuma and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use Kanuma?
Your doctor or nurse will give Kanuma to you or your child by an infusion (drip) into a vein.
More instructions can be found in Section 4. How do I use Kanuma? in the full CMI.
5. What should I know while using Kanuma?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using Kanuma? in the full CMI.
6. Are there any side effects?
Kanuma contains a protein and proteins can cause allergic reactions in some people including serious, potentially life-threatening allergic reaction. If you or your child experiences a severe infusion reaction like this, seek immediate medical attention.
The development of blood proteins against Kanuma, also called anti-drug antibodies, may occur during the treatment. Talk to your doctor if you experience decreased efficacy with Kanuma.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
KANUMA® (ka-nu-ma)
Active ingredient(s): [sebelipase alfa,rce]
Consumer Medicine Information (CMI)
This leaflet provides important information about using Kanuma.
You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using Kanuma.
Where to find information in this leaflet:
1. Why am I using Kanuma?
2. What should I know before I use Kanuma?
3. What if I am taking other medicines?
4. How do I use Kanuma?
5. What should I know while using Kanuma?
6. Are there any side effects?
7. Product details
1. Why am I using Kanuma?
Kanuma contains the active ingredient sebelipase alfa rce.
Sebelipase alfa rce is similar to the naturally occurring enzyme lysosomal acid lipase (LAL), which the body uses to breakdown fats.
Kanuma is an enzyme replacement therapy. This means that it replaces the missing or defective LAL enzyme in patients with LAL-D.
Kanuma is used to treat patients of all ages with lysosomal acid lipase deficiency (LAL-D).
Kanuma works by lowering the buildup of fat that causes medical complications, including impaired growth, digestive problems, liver damage, including liver failure and heart complications. It also improves blood levels of fats, including reducing elevated low-density lipoprotein cholesterol (LDL; also known as bad cholesterol) and triglycerides.
2. What should I know before I use Kanuma?
Warnings
Do not use Kanuma if you or your child has had a life-threatening allergic reaction:
- to sebelipase alfa rce,
- to any of the ingredients listed at the end of this leaflet, or
- to egg
that cannot be managed when you or your child receives Kanuma again.
Some symptoms of an allergic reaction may include:
- shortness of breath, wheezing or difficulty breathing,
- swelling of the face, lips, tongue, or other parts of the body,
- rash, itching or hives on the skin.
If you are not sure whether you or your child should be treated with Kanuma, talk to your doctor.
You or your child may experience side effects while being given Kanuma, or during the hours following the infusion. This is known as an infusion reaction. It can sometimes be severe and may include an allergic reaction (see above "Some symptoms of an allergic reaction may include"). If you or your child experiences a severe infusion reaction, seek immediate medical attention.
If you or your child has an infusion reaction you may be given additional medicines to treat or prevent future reactions. These medicines may include antihistamines, fever-reducing medicines or corticosteroids (a type of anti-inflammatory medicines).
If the infusion reaction is severe, your doctor may stop the infusion.
Check with your doctor if you:
- or your child has allergies to any other medicines, foods, preservatives, or dyes.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant, your doctor can discuss with you the risks and benefits involved. There is insufficient data from the use of Kanuma in pregnant women to determine if sebelipase alfa rce exposure during pregnancy poses any risk to the mother or fetus.
Talk to your doctor if you are breastfeeding or intend to breastfeed. It is not known whether Kanuma passes into breast milk. You should not breast feed while using Kanuma unless you have discussed it with your doctor.
Your doctor will discuss the benefits and risks of continuing treatment with Kanuma whilst breastfeeding.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you or your child are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
It is not known if Kanuma interferes with other medicines or other medicines affects Kanuma and how it works.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect Kanuma.
4. How do I use Kanuma?
Follow all directions given to you by your doctor, nurse or pharmacist carefully. They may differ from the information contained in this leaflet.
The dose you or your child receives is based on body weight.
Infants (less than 6 months of age)
For patients who have signs and symptoms of the disease when they are infants, the recommended starting dose is 1 mg per kg or 3 mg per kg of body weight once weekly.
Children and adults
The recommended dose is 1 mg per kg body weight once every two weeks.
Dose adjustments may be considered based on how well your child responds to treatment.
Kanuma should be started at as young an age as possible and is intended for long-term use.
Your doctor or nurse will give Kanuma to you or your child by an infusion (drip) into a vein. Kanuma will be diluted before being administered.
Each infusion will take approximately 1 to 2 hours. You or your child will be monitored by your doctor or nurse after infusion.
If you forget to use Kanuma
If you forget or miss you or your child's appointment for a Kanuma infusion, contact your doctor immediately.
If you use too much Kanuma
As Kanuma is given to you or your child under the supervision of your doctor, it is unlikely that you will receive too much.
5. What should I know while using Kanuma?
Things you should do
- Tell any other doctors, dentists, and pharmacists who treat you that you or your child is taking this medicine.
- Keep appointments with your doctor or clinic. It is important to have your or your child's Kanuma infusion at the appointed time each time to make sure Kanuma has the best chance of providing effective treatment for the condition.
Call your doctor straight away if you:
If you or your child experiences a severe infusion reaction.
You or your child may experience a side effect while you or your child is being given Kanuma or during the hours following the infusion.
This is known as an infusion reaction which can sometimes be severe and may include an allergic reaction that could be life-threatening and require medical treatment.
Things you should not do
- Do not stop using this medicine suddenly.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how Kanuma affects you.
It is not known if Kanuma impairs the ability to drive or operate machinery. However, if you experience dizziness, a reported side effect it could affect your ability to drive or operate machinery until it has resolved.
Looking after your medicine
Kanuma will be stored under refrigeration (2°C to 8°C, do not freeze) in the original packaging in order to protect from light in the hospital or pharmacy.
When to discard your medicine
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
Side effects reported in infants (1 to 6 months old) are:
Side effects reported in children and adults are:
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What Kanuma contains
| Active ingredient (main ingredient) | Sebelipase alfa rce |
| Other ingredients (inactive ingredients) | Sodium citrate Citric acid monohydrate Albumin Water for Injection |
| Potential allergens | Egg |
Do not take this medicine if you are allergic to any of these ingredients.
What Kanuma looks like
Kanuma is a clear to slightly opalescent, and colourless to slightly coloured solution contained in a glass vial
Pack size: 1 vial containing 10 mL of concentrate
(AUST R 274498).
Who distributes Kanuma
Alexion Pharmaceuticals Australasia Pty Ltd
Level 4
66 Talavera Road, Macquarie Park
NSW 2113
Medical enquiries: 1800 788 189
This leaflet was prepared in June 2023.
Brand Information
| Brand name | Kanuma |
| Active ingredient | Sebelipase alfa |
| Schedule | S4 |
MIMS Revision Date: 01 July 2023
1 Name of Medicine
Sebelipase alfa rce.
2 Qualitative and Quantitative Composition
Kanuma is supplied as a single-use vial containing 20 mg of sebelipase alfa rce (2 mg/mL).
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Concentrated solution for intravenous infusion. Kanuma is a clear to slightly opalescent, colourless to slightly coloured solution.
4 Clinical Particulars
4.1 Therapeutic Indications
Kanuma (sebelipase alfa rce) is indicated for long-term enzyme replacement therapy (ERT) in patients of all ages with lysosomal acid lipase deficiency (LAL-D).
4.2 Dose and Method of Administration
It is important to initiate treatment as early as possible.
Kanuma is for intravenous use only. The total volume of the infusion should be administered over approximately 2 hours. Infusion over 1 hour may be considered for those patients receiving the 1 mg/kg dose after patient tolerability is established. The infusion period may be extended in the event of dose escalation.
For instructions on the preventive measures and monitoring of hypersensitivity reactions, see Section 4.4 Special Warnings and Precautions for Use, Hypersensitivity reactions, including anaphylactic reactions or anaphylaxis.
Recommended dose. Patients with rapidly progressive LAL deficiency presenting within the first 6 months of life. The recommended starting dose in infants (< 6 months of age) presenting with rapidly progressive LAL-D is 1 mg/kg or 3 mg/kg administered as an IV infusion once weekly depending on the clinical status of the patient.
Dose escalations may be considered based on suboptimal response to clinical and biochemical criteria, including, poor growth, deteriorating biochemical markers (e.g. liver transaminases, ferritin, C-reactive protein, and coagulation parameters), persistent or worsening organomegaly, increased frequency of intercurrent infections, and persistent or worsening of other symptoms (e.g. gastrointestinal symptoms).
A dose escalation to 3 mg/kg should be considered in case of suboptimal clinical response.
A further dose escalation up to 5 mg/kg may be considered in case of persistent suboptimal clinical response.
Further dose adjustments, as a reduction of the dose or an extension of the dose interval, can be made on an individual basis based on achievement and maintenance of therapeutic goals. Clinical studies evaluated doses ranging from 0.35 to 5 mg/kg once weekly, with 1 patient receiving a higher dose of 7.5 mg/kg once weekly (see Section 5.1 Pharmacodynamic Properties). Doses higher than 7.5 mg/kg have not been studied.
Paediatric patients and adults presenting with LAL-D. The recommended dose in children and adults presenting with LAL-D is 1 mg/kg administered every two weeks as an IV infusion. Dose escalation to 3 mg/kg once every two weeks could be considered based on suboptimal response to clinical and biochemical criteria, including, poor growth, persistent or deteriorating biochemical markers (e.g. liver transaminases, parameters of lipid metabolism), persistent or worsening organomegaly, and persistent worsening of other symptoms (e.g. gastrointestinal symptoms).
Method of administration. It is recommended to allow Kanuma vials to reach a temperature between 15°C and 25°C prior to reconstitution to minimize the potential for the formation of sebelipase alfa rce protein particles in solution.
Dilute Kanuma with 0.9% sodium chloride solution for infusion using aseptic technique. The diluted solution should be administered to patients using a low-protein binding infusion set equipped with an in-line, low-protein binding 0.2 micrometre filter with a surface area of greater than 4.5 cm2 as available, in order to avoid filter occlusion.
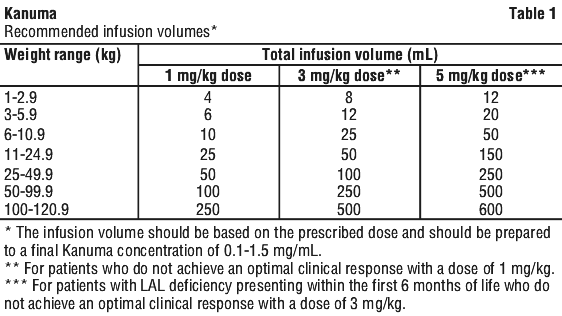
Preparation of the Kanuma infusion. Determine the number of vials to be diluted for infusion based on the patient's weight and prescribed dose.
Dilute the total calculated dose with 0.9% sodium chloride solution for infusion. See Table 1 for recommended infusion volumes by weight range.
Product is for single use in one patient only. Discard any unused portion left in the vial, as the product contains no preservatives.
To reduce microbiological hazard, use as soon as practicable after dilution. If storage is necessary, hold at 2°-8°C for not more than 24 hours or up to 8 hours below 25°C.
Special populations. Patients with renal and hepatic impairment. No dosing adjustment is recommended in patients with renal or hepatic impairment based on current knowledge of the pharmacokinetics and pharmacodynamics of sebelipase alfa rce.
Adult patients. Safety and efficacy data in patients > 18 years old are limited.
LAL-D patient monitoring program. Physicians are encouraged to participate and enrol all patients diagnosed with LAL-D in a patient monitoring program.
4.3 Contraindications
Kanuma is contraindicated in patients with a life-threatening hypersensitivity (anaphylactic reaction) to the active substance, to egg, or to any of the excipients (see Section 6.1 List of Excipients), when attempts to rechallenge are unsuccessful.
4.4 Special Warnings and Precautions for Use
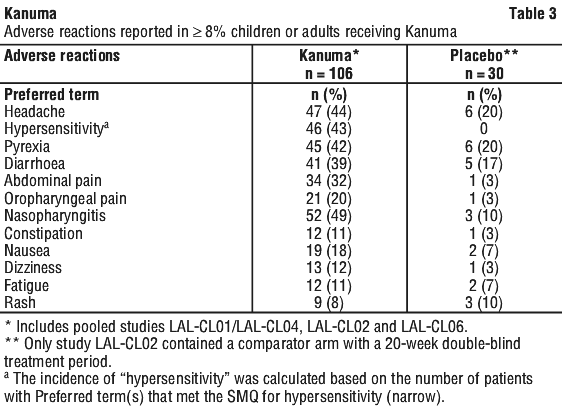
Hypersensitivity reactions, including anaphylactic reactions or anaphylaxis. Hypersensitivity reactions, including anaphylactic reactions or anaphylaxis, have been reported in Kanuma-treated patients. In clinical trials, 5 of 125 (4%) patients treated with Kanuma, including 3 of 19 (16%) infants and 2 of 106 (2%) children and adults, experienced serious signs and symptoms consistent with anaphylaxis. These patients experienced reactions during infusion with signs and symptoms including chest discomfort, conjunctival hyperaemia, dyspnoea, hyperaemia, eyelid oedema, rhinorrhoea, severe respiratory distress, tachycardia, tachypnoea, irritability, flushing, pruritus, stridor, hypoxia, pallor, diarrhoea and urticaria. Anaphylaxis has occurred as late as 1 year after treatment initiation.
In clinical trials, 59 of 125 (47%) Kanuma-treated patients, including 13 of 19 (68%) infants and 46 of 106 (43%) paediatric patients, 4 years and older, and adults, experienced at least 1 hypersensitivity reaction (selected using a validated, predetermined set of terms grouped together to identify potential hypersensitivity reactions). Signs and symptoms either consistent with, or that may be related to a hypersensitivity reaction occurring in 2 or more patients, included but were not limited to abdominal pain, agitation, bronchospasm, fever, chills, diarrhoea, eyelid oedema, eczema, face oedema, hypertension, irritability, laryngeal oedema, lip swelling, nausea, oedema, pallor, pruritus, rash, tachycardia, urticaria and vomiting. The majority of reactions occurred during or within 4 hours of the completion of the infusion. Patients were not routinely pre-medicated prior to infusion of Kanuma in these clinical trials.
Appropriate medical support should be readily available when Kanuma is administered. If anaphylaxis occurs, immediately discontinue the infusion and initiate appropriate medical treatment. Patients should be closely observed during, and after the infusion. Inform patients of the signs and symptoms of anaphylaxis, and instruct them to seek immediate medical care should signs and symptoms occur.
The risks and benefits of re-administering Kanuma following a severe reaction should be considered. For patients who have experienced allergic reactions during infusion, caution should be exercised upon re-administration.
The management of hypersensitivity reactions should be based on the severity of the reaction and may include temporarily interrupting the infusion, lowering the infusion rate, and/or treatment with antihistamines, antipyretics, and/or corticosteroids. If interrupted, the infusion may be resumed at a slower rate with increases as tolerated. Pre-treatment with antipyretics and/or antihistamines may prevent subsequent reactions in those cases where symptomatic treatment was required.
Hypersensitivity to egg. Sebelipase alfa rce is produced in the egg white of transgenic Gallus by recombinant DNA technology. Patients with a known history of egg allergies were excluded from the clinical trials. Consider the risks and benefits of treatment with Kanuma in patients with known life-threatening hypersensitivity (anaphylactic reaction) to egg.
Immunogenicity. As with all therapeutic proteins, there is potential for immunogenicity. In the Kanuma clinical program, patients were routinely tested for anti-sebelipase alfa rce anti-drug antibodies (ADAs) to determine the immunogenicity potential of sebelipase alfa rce. Patients who tested positive for ADAs were also tested for inhibitory antibody activity. The presence of inhibitory antibody activity has been detected at some post-baseline timepoints in clinical studies (see Section 4.8 Adverse Effects (Undesirable Effects)).
Overall, there is no clear relationship between either development of ADAs/inhibitory antibody activity and associated hypersensitivity reactions or suboptimal clinical response.
During clinical trials, a decrease in clinical response associated with the development of inhibitory antibody activity was only observed in 3 patients with a homozygous deletion affecting both alleles of genes Lipase A, lysosomal acid (LIPA) and Cholesterol, 25-Hydroxylase (see Section 4.8 Adverse Effects (Undesirable Effects)).
Paediatric use. Eighty-eight of 125 patients (70%) who received Kanuma during clinical studies were in the paediatric and adolescent age range (0.5 months up to 17.6 years) at the time of first dose.
Use in the elderly. Safety and efficacy of sebelipase alfa rce in patients older than 65 years have not been established. Therefore, there is no information available to determine whether patients aged 65 years and over respond differently from younger patients.
Effects on laboratory tests. Unknown.
4.5 Interactions with Other Medicines and Other Forms of Interactions
Drug interaction studies have not been performed with sebelipase alfa rce.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. No adverse effects on fertility and reproductive performance were observed in male and female rats given IV doses of sebelipase alfa rce up to 60 mg/kg administered twice weekly (approximately 122 times the adult human AUC of 1861 nanogram.h/mL at 1 mg/kg dose administered every two weeks).
Use in pregnancy. (Category B1)
Sebelipase alfa rce administered during the period of organogenesis to rats (on gestation days 6, 9, 12, 15 and 17) and rabbits (on gestation days 7, 10, 13, 16 and 19) at intravenous doses up to 60 and 50 mg/kg, respectively (approximately 122 and 392 times the human AUC of 1861 nanogram.h/mL at 1 mg/kg dose administered every two weeks, respectively) did not cause any adverse effects on embryofoetal development. A pre- and postnatal development study in rats showed no evidence of adverse effects on pre- and postnatal development at intravenous doses (administered on gestation days 6, 9, 12, 15, 18, and 20 and days 4, 7, 10, 14, and 17 postpartum) of sebelipase alfa rce up to 60 mg/kg/day (approximately 122 times the human AUC of 1861 nanogram.h/mL at 1 mg/kg dose administered every two weeks).
There is insufficient data from the use of Kanuma in pregnant women to determine if sebelipase alfa rce exposure during pregnancy poses any risk to the mother or fetus. Pregnant and lactating women were excluded from Kanuma clinical trials. As a precautionary measure, it is preferable to avoid the use of Kanuma during pregnancy.
Use in lactation. There are no data from studies in breast-feeding women. It is not known whether Kanuma is excreted in human milk. A decision must be made whether to discontinue breast-feeding or to discontinue/abstain from Kanuma treatment taking into account the benefit of breast-feeding for the child and the benefit of therapy for the woman.
4.7 Effects on Ability to Drive and Use Machines
No specific studies have been conducted to assess the direct effect of Kanuma on the ability to drive and use machines. However, adverse effects of Kanuma include dizziness which could affect the ability to drive or use machines. See Section 4.8 Adverse Effects (Undesirable Effects).
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile. The data described below reflect exposure of Kanuma in 125 patients who received Kanuma at dosages ranging from 0.35 mg/kg every two weeks to 7.5 mg/kg weekly in clinical studies. Patients were between 0.5 months and 59 years old (70% were < 18 years old) at the time of first treatment with Kanuma.
In infants, the safety of Kanuma was evaluated in an open-label, single arm Phase 2/3 study (Study LAL-CL03; weekly doses of 0.35, 1, 3, and 5 mg/kg), see Section 5.1, Clinical trials) and in an open-label, Phase 2 study (Study LAL-CL08; starting dose of 1 mg/kg weekly with potential sequential dose escalations when dose escalation criteria were met, to 3 mg/kg weekly, 5 mg/kg weekly, and 7.5 mg/kg weekly). Fifteen of nineteen infants received dose escalations as allowed by the study protocol from 1 mg/kg to 3 mg/kg administered weekly. Among these patients, nine patients had a further dose escalation to 5 mg/kg weekly, and one of these patients received a further dose escalation to 7.5 mg/kg weekly. Four of nineteen infants died for reasons unrelated to Kanuma prior to a protocol-defined dose escalation to 3 mg/kg once weekly.
In children and adults, the safety of Kanuma was evaluated in a randomised placebo-controlled Phase 3 study (Study LAL-CL02; doses of 1 mg/kg every two weeks during the double blind phase and doses of 1 and 3 mg/kg every two weeks during the extension phase); an open-label, single-arm, Phase 1/2 study in adults (Study LAL-CL01; doses of 0.35 mg, 1 mg, or 3 mg/kg weekly for four weeks, followed by a period when patients were off treatment with Kanuma before entering an extension period (Study LAL-CL04) during which patients resumed the previous dosage for another four weeks and then switched to a regimen of 1 or 3 mg/kg every two weeks; and an open-label, Phase 2 study in children and adults (≥ 8 months) (Study LAL-CL06; dose of 1 mg/kg every two weeks, with potential dose escalations up to 3 mg/kg weekly when subjects met dose escalation criteria). Twenty-three of 106 children and adults received dose escalations as allowed by the study protocol from 1 mg/kg to 3 mg/kg administered every two weeks, and four of these patients had a further dose escalation to 3 mg/kg weekly.
Overall, 102/106 (96.2%) children and adult patients have received Kanuma administered at a dosage regimen of 1 mg/kg every 2 weeks, with a median duration of exposure of 33 months (6,59 months). The majority of these 102 patients were from Study LAL-CL02 (66/102, 64.7%). The median duration of exposure for the nineteen infants enrolled in clinical trials was 35.6 months (1 day, 60 months).
The most serious adverse reactions experienced by 4% of patients in clinical trials were signs and symptoms consistent with anaphylaxis, see Section 4.4 Special Warnings and Precautions for Use, Hypersensitivity reactions, including anaphylactic reactions or anaphylaxis.
Tabulated list of adverse reactions. Table 2 summarises the most common adverse reactions occurring in ≥ 30% of patients with rapidly progressive LAL-D presenting in the first 6 months of life receiving Kanuma.

Immune system disorder. Anaphylactic reactionb.
Eye disorder. Eyelid oedema.
Skin and subcutaneous disorders. Rash maculo-papular.
General disorders and administration site conditions. Hyperthermia.
Investigations. Body temperature increased, respiratory rate increased, heart rate increased, blood pressure increased, drug specific antibody present, oxygen saturation decreased.
b Occurred in 3 infant patients treated in clinical trials. Based on Preferred term 'anaphylactic reaction' and application of Sampson criteria to identify signs/symptoms consistent with anaphylaxis.
Adverse reactions that occurred in children or adults at rates less than 8% included (presented by SOC [italicized] and PT). Immune system disorder. Anaphylactic reactionc.
Cardiac disorder. Tachycardia.
Vascular disorders. Hyperaemia, hypotension.
Respiratory, thoracic and mediastinal disorders. Dyspnoea.
Gastrointestinal disorders. Abdominal distention.
Skin and subcutaneous disorders. Rash, rash papular.
General disorders and administration site conditions. Asthenia, chest discomfort, infusion site reaction.
Investigations. Body temperature increased.
c Occurred in 2 patients treated in clinical trials. Based on Preferred term 'anaphylactic reaction' and application of Sampson criteria to identify signs/symptoms consistent with anaphylaxis.
Description of selected adverse reactions. Transient hyperlipidaemia. Consistent with its known mechanism of action, asymptomatic increases in circulating cholesterol and triglycerides have been observed following initiation of Kanuma. These increases have generally occurred within the first 2 to 4 weeks and improved within a further 8 weeks of Kanuma treatment.
Immunogenicity. There is potential for immunogenicity, (see Section 4.4 Special Warnings and Precautions for Use).
Among 106 children and adults with LAL deficiency enrolled in the clinical studies, 9/106 (8%) patients were reported with ADA positivity and 2/106 (2%) of patients tested positive for NAbs that inhibited enzyme activity and/or cellular uptake. Among 19 infants with LAL deficiency enrolled in the clinical studies, 10/19 (53%) patients were reported with ADA positivity and 9/19 (47%) patients tested positive for NAbs that inhibited enzyme activity and/or cellular uptake.
Overall, there is no clear relationship between development of ADAs/NAbs and associated hypersensitivity reactions or suboptimal clinical response. In clinical studies, 3 patients homozygous for a deletion affecting both alleles of genes Lipase A, lysosomal acid [LIPA] and Cholesterol 25-Hydroxylase developed inhibitory antibody activity associated with a suboptimal clinical response (see Section 4.4 Special Warnings and Precautions for Use). These patients underwent either immunomodulatory therapy alone or in combination with hematopoietic stem cell transplant (HSCT) or bone marrow transplant (BMT), resulting in improved clinical response to Kanuma.
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
4.9 Overdose
The maximum dose of Kanuma used in clinical trials was 7.5 mg/kg/week. No specific signs or symptoms were identified following the higher doses.
For information on the management of overdose, contact the Poison Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action. Lysosomal acid lipase deficiency (LAL-D) is a rare disease associated with significant morbidity and mortality affecting individuals from infancy through to adulthood. LAL-D presenting in infants is a medical emergency with rapid disease progression over a period of weeks that is typically fatal within the first 6 months of life.
LAL-D is an autosomal recessive lysosomal storage disorder characterised by a genetic defect resulting in a marked decrease or loss in activity of the lysosomal acid lipase (LAL) enzyme. Deficient LAL enzyme activity results in progressive complications due to the lysosomal accumulation of cholesteryl esters and triglycerides in multiple organs, including the liver, spleen, intestine, and the walls of blood vessels. The resulting lipid accumulation in the liver leads to hepatomegaly, increased hepatic fat content, transaminase elevation signaling chronic liver injury, and progression to fibrosis, cirrhosis, and complications of end stage liver disease. In the spleen, LAL deficiency results in splenomegaly, anaemia, and thrombocytopenia. Lipid accumulation in the intestinal wall leads to malabsorption and growth failure. In parallel, dyslipidemia due to impaired degradation of lysosomal lipid is common with elevated low-density lipoprotein cholesterol (LDL-c) and triglycerides, and low high-density lipoprotein cholesterol (HDL-c). In addition to liver disease, patients with LAL-D experience increased risk for cardiovascular disease and accelerated atherosclerosis.
Sebelipase alfa rce is taken up by cells and is subsequently internalised into lysosomes where it catalyzes the lysosomal hydrolysis of cholesteryl esters and triglycerides to free cholesterol, glycerol and free fatty acids. Binding of glycans present on sebelipase alfa rce, to cell surface receptors is implicated in the cellular uptake of sebelipase alfa rce. Treatment with sebelipase alfa rce restores LAL enzyme activity in LAL-D cells, enabling hydrolysis of cholesteryl esters and triglycerides in the lysosome. Sebelipase alfa rce treatment restored hepatic LAL activity and lead to reductions in the fat content of the liver and spleen; reductions in serum transaminases, LDL-c, non-HDL-c, and triglycerides; and increases in serum HDL-c. Improvement in growth was associated with LAL substrate reduction in the intestine.
Pharmacodynamics. In clinical trials, after initiation of dosing with Kanuma, breakdown of accumulated lysosomal lipid led to initial increases in LDL-c and triglycerides within the first 2 to 4 weeks of treatment. In general, following increases in LDL-c and triglycerides, these parameters decreased to below pre-treatment values within 8 weeks of treatment with Kanuma.
In all patients with elevated alanine aminotransferase (ALT) values at baseline (82 of 84 patients in clinical trials), reductions in ALT values were observed, generally within 2 weeks after initiation of treatment with Kanuma. In open-label studies, some infants showed sustained improvement for up to 260 weeks, and some children and adults showed sustained improvement for up to 256 weeks. Treatment interruption resulted in increases in LDL-c and ALT values and decreases in HDL-c.
Clinical trials. Infants presenting with LAL deficiency. Study LAL-CL03. Study LAL-CL03 was a multicentre, open-label, single-arm study of Kanuma in 9 patients with LAL-D with growth failure, or other evidence of rapidly progressive disease prior to 6 months of age. Patients had rapidly progressive liver disease and severe hepatosplenomegaly. The age range at study entry was 1-6 months. Patients received Kanuma at 0.35 mg/kg per week for the first 2 weeks, and then 1 mg/kg per week. Based on clinical response, dose escalation to 3 mg/kg per week occurred as early as 1 month and up to 20 months after starting treatment at 1 mg/kg. A further dose escalation to 5 mg/kg per week was allowed.
Efficacy was assessed by comparing the survival experience of Kanuma-treated patients who survived past 12 months of age in study LAL-CL03 with a historical cohort of untreated infants presenting with LAL-D with similar clinical characteristics. In study LAL-CL03, 6 of the 9 Kanuma-treated infants survived beyond 12 months (67% 12-month survival; 95% CI: 30% to 93%). With continued treatment beyond 12 months of age, 1 additional patient died at age 15 months. In the historical cohort, 0 of 21 patients survived beyond 8 months of age (0% 12-month survival, 95% CI: 0% to 16%).
Kanuma at doses up to 1 mg/kg per week resulted in improvements in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels and weight gain within the first several weeks of treatment. From baseline to week 48, the mean reductions for ALT and AST were -34.0 U/L and -44.5 U/L, respectively. From baseline to week 48, mean weight-for-age percentile improved from 12.74% to 29.83% and mean serum albumin levels increased from 26.7 g/L to 38.7 g/L. Dose escalation to 3 mg/kg per week was associated with additional improvements in weight gain, lymphadenopathy and serum albumin.
Children and adults with LAL deficiency. Study LAL-CL02. Study LAL-CL02 was a multicentre, double-blind, placebo-controlled study in 66 children and adults with LAL-D. Patients were randomised to receive a Kanuma dose of 1 mg/kg (n = 36) or placebo (n = 30) every two weeks for 20 weeks in the double-blind period. The median age at randomisation was 13.5 years, range 4-58 years, (36% were < 12 years old and 71% were < 18 years old). For study entry, patients were required to have ALT levels of ≥ 1.5 x upper limit of normal (ULN). The majority of patients (58%) had LDL-c > 4.91 mmol/L at study entry, and 24% of patients with LDL-c > 4.91 mmol/L were on lipid lowering medications. Of the 32 patients who had a liver biopsy at study entry, 100% had fibrosis and 31% had cirrhosis. The age range of patients with biopsy evidence of cirrhosis was 4-21 years.
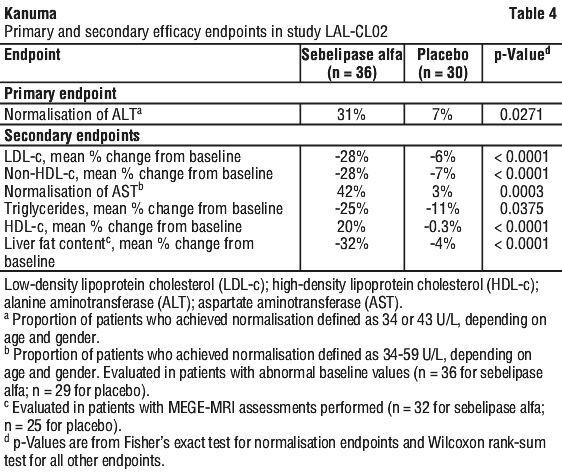
Randomisation was stratified by 1) age at randomisation (< 12 years old; ≥ 12 years old); 2) ALT level at screening (< 3 x ULN; ≥ 3 x ULN); and 3) use of lipid lowering medications (yes; no). The following endpoints were assessed: normalisation of ALT, decrease in LDL-c, decrease in non-HDL-c, normalisation of AST, decrease in triglycerides, increase in HDL-c, decrease in liver fat content assessed by multi-echo gradient echo magnetic resonance imaging (MEGE-MRI), and improvement in hepatic steatosis measured by morphometry.
Through an exploratory analysis, a statistically significant improvement in multiple endpoints was observed in the Kanuma-treated group as compared to the placebo group at the completion of the 20-week double-blind period, as shown in Table 4. Normalisation of ALT was achieved in 31% (11/36) of Kanuma-treated patients and 7% (2/30) of placebo patients. LDL-c normalisation (< 3.36 mmol/L) was achieved in 40.6% (13/32) of Kanuma-treated patients and 6.7% (2/30) of placebo patients with abnormal baseline LDL-c (≥ 3.36 mmol/L).
Patients treated with Kanuma had larger reductions from baseline in ALT values and liver fat content (measured by MRI) compared to patients treated with placebo. The absolute reduction in mean ALT level was -57.9 U/L (-53%) in the Kanuma-treated group and -6.7 U/L (-6%) in the placebo group.
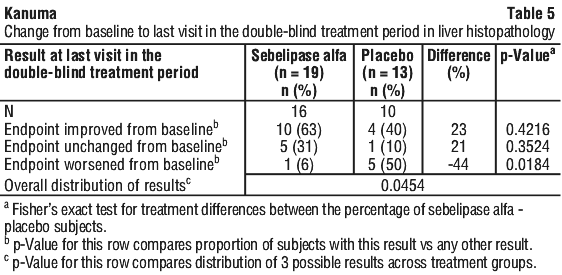
Placebo patients had persistently elevated serum transaminase and abnormal serum lipid levels during the double-blind period. Consistent with what was observed in Kanuma-treated patients during the double-blind period, initiation of treatment with Kanuma during the open-label period produced rapid improvements in ALT levels and in lipid parameters including LDL-c and HDL-c levels (see Figure 1 and Figure 2).
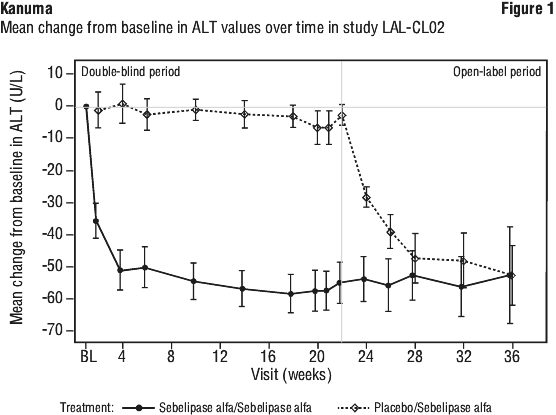
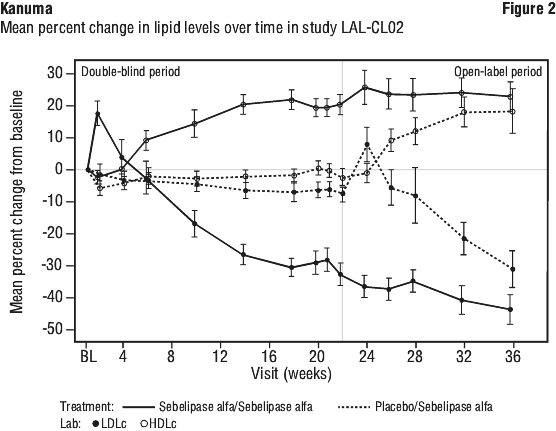
5.2 Pharmacokinetic Properties
No conclusion on the linearity of sebelipase alfa rce pharmacokinetics can be made due to limited data at higher exposures. No drug accumulation is observed following 1 mg/kg or 3 mg/kg once every other week dosing, although observations for the drug accumulation at 3 mg/kg every other week are based on a limited number of patients. Accumulation following once weekly dosing is not expected based on relatively rapid drug clearance.
The pharmacokinetics of sebelipase alfa rce in children and adults were determined using a population pharmacokinetic analysis of 102 patients with LAL-D who received intravenous infusions of Kanuma across 4 clinical studies (LAL-CL02, LAL-CL03, LAL-CL04 and LAL-CL06 see Table 6). Four patients were below 2 years old, 33 were 2 to 11 years old, 34 were 12 to 17 years old, and 31 were adults (18 years and over). The pharmacokinetic profiles of sebelipase alfa rce were similar between adolescents and adults. The T1/2 was similar across all age groups.
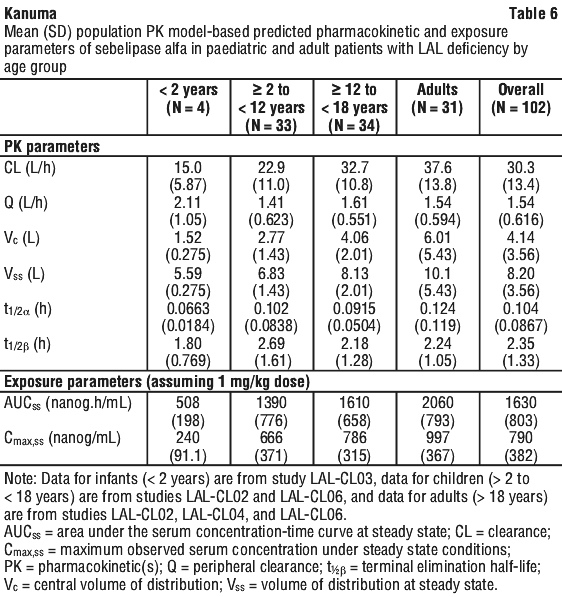
Special populations. During the covariate analysis of the population pharmacokinetics model for sebelipase alfa rce, age, sex, and enzyme maturation were found not to have a significant influence on CL (drug clearance) and Vc (central volume of distribution) of sebelipase alfa rce. Body weight and body surface area are significant covariates on CL. Sebelipase alfa rce has not been investigated in patients 65 years of age or older.
There is limited information on the impact of anti-drug antibodies on sebelipase alfa rce pharmacokinetics. As with all therapeutic proteins, there is the potential for the development of immunogenicity. Nineteen of 125 (15%) patients with LAL deficiency had at least 1 post-baseline antidrug antibody (ADA) positive result, 10 of which were infants. Among children and adult patients with LAL deficiency, ADA positivity was transient with generally low titres of ADAs reported. In the absence of additional treatment, e.g. immunomodulation, ADA positivity appeared to be persistent infants. There was no observed impact of anti-drug antibody development on sebelipase alfa rce pharmacokinetics.
Renal and hepatic impairment. Sebelipase alfa rce is expected to be metabolically degraded through peptide hydrolysis. Consequently, impaired liver function is not expected to affect the pharmacokinetics of sebelipase alfa rce. There is a lack of data in patients with severe hepatic impairment.
Renal elimination of sebelipase alfa rce is considered a minor pathway for clearance. There is a lack of data in patients with renal impairment.
5.3 Preclinical Safety Data
Genotoxicity. No studies have been conducted to assess the genotoxic potential of sebelipase alfa rce.
Carcinogenicity. No studies have been conducted to assess the carcinogenic potential of sebelipase alfa rce.
6 Pharmaceutical Particulars
6.1 List of Excipients
Sodium citrate, citric acid monohydrate, albumin, water for injections.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
Do not use beyond the expiration date (EXP) stamped on the packaging.
6.4 Special Precautions for Storage
Kanuma vials must be stored in a refrigerator (2 to 8°C, Do not freeze) in the original packaging in order to protect from light.
6.5 Nature and Contents of Container
Single unit 20 mg carton. Contains one 10 mL vial.
6.6 Special Precautions for Disposal
Product is for single use in one patient only. Discard any unused portion left in the vial, as the product contains no preservatives. Unused or expired medicine should be returned to a pharmacy for disposal.
6.7 Physicochemical Properties
Chemical structure.
Sebelipase alfa rce is a recombinant human lysosomal acid lipase (rhLAL) produced by recombinant DNA technology and purified from the egg white of genetically engineered chickens {transgenic Gallus}. Purified sebelipase alfa rce is a monomeric glycoprotein containing 6 N-linked glycosylation sites with a molecular weight of approx. 55 kDa.
7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.
Date of First Approval
18 May 2017
Date of Revision
06 June 2023
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.