Lamivudine Viatris
Brand Information
| Brand name | Lamivudine Viatris |
| Active ingredient | Lamivudine |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Lamivudine Viatris.
Summary CMI
LAMIVUDINE VIATRIS
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about taking LAMIVUDINE VIATRIS, speak to your doctor or pharmacist.
1. Why am I taking LAMIVUDINE VIATRIS?
LAMIVUDINE VIATRIS contains the lamivudine. LAMIVUDINE VIATRIS are used to slow down the progression of human immunodeficiency virus (HIV) infection.
For more information, see Section 1. Why am I taking LAMIVUDINE VIATRIS? in the full CMI.
2. What should I know before I take LAMIVUDINE VIATRIS?
Do not take LAMIVUDINE VIATRIS if you have ever had an allergic reaction to lamivudine or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other health problems, take any other medicines, are pregnant or breastfeeding, or plan to become pregnant or breastfeed.
For more information, see Section 2. What should I know before I take LAMIVUDINE VIATRIS? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with LAMIVUDINE VIATRIS and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I take LAMIVUDINE VIATRIS?
- The usual dosage of LAMIVUDINE VIATRIS is one 150 mg tablet twice a day or 300 mg once a day. For younger children between 3 months to 12 years of age the dose of LAMIVUDINE VIATRIS will depend on their weight in kilograms (kg).
- Swallow the tablet whole with a glass of water.
- Your doctor may prescribe a different dosage or provide additional instructions.
More instructions can be found in Section 4. How do I take LAMIVUDINE VIATRIS? in the full CMI.
5. What should I know while taking LAMIVUDINE VIATRIS?
| Things you must do |
|
| Things you must not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while taking LAMIVUDINE VIATRIS? in the full CMI.
6. Are there any side effects?
Common side effects that have been reported include nausea, vomiting, diarrhoea, upper abdominal pain, headache, high temperature, lethargy, fatigue, hair loss, joint and muscle pain, skin rash and increased bruising or bleeding. Serious side effects include lumpy skin rash or "hives", swelling of the face, lips, mouth, or throat, wheezing, chest pain or tightness, and fainting, which may indicate an allergic reaction. Severe stomach pain or cramps, nausea or vomiting may indicate pancreatitis.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
LAMIVUDINE VIATRIS
Active ingredient(s): lamivudine
Consumer Medicine Information (CMI)
This leaflet provides important information about taking LAMIVUDINE VIATRIS. Speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about taking LAMIVUDINE VIATRIS.
Where to find information in this leaflet:
1. Why am I taking LAMIVUDINE VIATRIS?
2. What should I know before I take LAMIVUDINE VIATRIS?
3. What if I am taking other medicines?
4. How do I take LAMIVUDINE VIATRIS?
5. What should I know while taking LAMIVUDINE VIATRIS?
6. Are there any side effects?
7. Product details
1. Why am I taking LAMIVUDINE VIATRIS?
LAMIVUDINE VIATRIS contain the active ingredient lamivudine.
Lamivudine belongs to a group of medicines called antivirals.
LAMIVUDINE VIATRIS are used together with other antivirals to slow down the progression of human immunodeficiency virus (HIV), which can lead to Acquired Immune Deficiency Syndrome (AIDS) and other related illnesses (e.g. AIDS-related Complex or ARC).
LAMIVUDINE VIATRIS do not cure AIDS or kill the HIV virus but prevents further damage to the immune system by stopping production of new viruses.
Your doctor may have prescribed LAMIVUDINE VIATRIS for another reason.
Ask your doctor if you have any questions about why LAMIVUDINE VIATRIS tablets have been prescribed for you.
2. What should I know before I take LAMIVUDINE VIATRIS?
Warnings
Do not take LAMIVUDINE VIATRIS if:
- you are allergic to lamivudine, or any of the ingredients listed at the end of this leaflet.
Always check the ingredients to make sure you can take LAMIVUDINE VIATRIS. - the expiry date printed on the pack has passed as the tablets may not work as well.
- the packaging is torn or shows signs of tampering.
Check with your doctor if you:
- are allergic to foods, dyes, preservatives, or any other medicines.
- you have, or have ever had, liver problems, for example jaundice, hepatitis, virus affecting the liver, enlarged liver or liver scarring (cirrhosis) or if you have any risk factors for liver problems, e.g. excessive alcohol intake, illegal intravenous drug use with shared equipment, iron or copper storage disorders.
- you have, or have ever had, kidney problems.
- you have, or have ever had, problems with your pancreas.
- you have diabetes.
- you have any other illness, including those that you think are not related to HIV infection.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Tell your doctor if you are pregnant or plan to become pregnant.
Tell your doctor if you are breastfeeding or plan to breastfeed.
Your doctor will discuss the risks and benefits of using LAMIVUDINE VIATRIS if you are pregnant or breastfeeding.
If you stop taking LAMIVUDINE VIATRIS
If you have a long-standing viral infection of your liver (hepatitis B) it may flare up. This can cause serious illness particularly if your liver is already not working very well. If you have both HIV and hepatitis B, when you stop taking LAMIVUDINE VIATRIS, your doctor is likely to arrange tests from time to time to check how well your liver is working and to measure virus levels.
LAMIVUDINE VIATRIS are not addictive.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interfere with LAMIVUDINE VIATRIS. This may affect how they work and/or cause more side effects.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect LAMIVUDINE VIATRIS.
4. How do I take LAMIVUDINE VIATRIS?
How much and when to take LAMIVUDINE VIATRIS
- The usual dosage of LAMIVUDINE VIATRIS in adults and adolescents 12 years and older is one 150 mg tablet twice a day or 300 mg once a day. Your doctor may prescribe a different dosage.
- For younger children between 3 months to 12 years of age the dose of LAMIVUDINE VIATRIS will depend on their weight in kilograms (kg). If you are giving LAMIVUDINE VIATRIS to a child, follow your doctor's instructions.
- Follow the instructions provided and use LAMIVUDINE VIATRIS until your doctor tells you to stop.
How to take LAMIVUDINE VIATRIS
- Tablets should be swallowed whole with a glass of water.
- If you cannot swallow the tablet(s), you may crush and combine them with a small amount of food or drink and take all of the dose immediately.
How long to take LAMIVUDINE VIATRIS
- LAMIVUDINE VIATRIS helps to control your condition, but does not cure it, you will need to take the tablets every day. Do not stop taking your medicine without first talking to your doctor.
If you forget to take LAMIVUDINE VIATRIS
LAMIVUDINE VIATRIS should be used regularly at the same time each day. If you miss your dose at the usual time, take it as soon as you remember, then go back to taking it as you would normally.
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Do not take a double dose to make up for the dose you missed.
If you take too many LAMIVUDINE VIATRIS
If you think that you or anyone else has taken too many LAMIVUDINE VIATRIS, urgent medical attention may be needed.
You must immediately:
- phone the Poisons Information Centre
(by calling 13 11 26) for advice, or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
Do this even if there are no signs of discomfort or poisoning.
5. What should I know while taking LAMIVUDINE VIATRIS?
Things you must do
- Tell your doctor if, for any reason, you have not taken your medicine exactly as prescribed.
Otherwise, your doctor may think it was not effective and change your treatment unnecessarily. - Use proper precautions to prevent passing on HIV virus.
You can still pass on the virus when taking LAMIVUDINE VIATRIS, by sexual activity or by contamination with infected blood, although the risk is lowered by effective antiretroviral therapy. Discuss with your doctor the precautions needed to avoid infecting other people. - Keep in regular contact with your doctor.
While taking LAMIVUDINE VIATRIS and/or any other therapy for HIV disease, you may continue to develop other infections and other complications of HIV infection.
Call your doctor straight away if you:
- Become pregnant or are trying to become pregnant.
- Generally feel unwell with a loss of appetite, nausea, vomiting, itching, yellowness of the skin or eyes or dark coloured urine, or if the blood tests of your liver function are abnormal. It is likely you will have to stop taking LAMIVUDINE VIATRIS tablets.
- Have any problems while taking LAMIVUDINE VIATRIS, even if you do not think the problems relate to LAMIVUDINE VIATRIS or are not listed in this leaflet.
Tell your doctor, dentist or pharmacist you visit that you are taking LAMIVUDINE VIATRIS.
Things you must not do
- Do not stop taking LAMIVUDINE VIATRIS suddenly or change the dose without first checking with your doctor.
- Do not give LAMIVUDINE VIATRIS to anyone else, even if their symptoms seem similar to yours.
- Do not take LAMIVUDINE VIATRIS to treat any other complaints unless your doctor says to.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how LAMIVUDINE VIATRIS affects you.
LAMIVUDINE VIATRIS may cause lethargy or fatigue in some people.
Drinking alcohol
Tell your doctor if you drink alcohol.
Looking after your medicine
- Keep LAMIVUDINE VIATRIS in the bottle with the cap tightly closed until you take them.
- If you take LAMIVUDINE VIATRIS out of their pack, they may not keep well.
- Heat and dampness can destroy some medicines.
Follow the instructions on the carton on how to take care of your medicine.
Store it in a cool dry place where it stays below 30°C, away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Keep it where young children cannot see or reach it.
A locked cupboard at least one-and a-half meters above the ground is a good place to store medicines.
When to dispose of your medicine
- If you no longer need to take it
- After the expiry date
Getting rid of any unwanted medicine
Take it to any pharmacy for safe disposal.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
Gastrointestinal:
| Speak to your doctor if you have any of these less serious side effects. Should any change in body shape be noticed, seek medical advice. |
Serious side effects
| Serious side effects | What to do |
Immune system:
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Very Serious side effects
| Very Serious side effects | What to do |
Allergic reaction:
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these very serious side effects. Do not take any more LAMIVUDINE VIATRIS. All these side effects are very rare. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Some people may have side effects that are not listed here.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
LAMIVUDINE VIATRIS is only available with a doctor's prescription.
What LAMIVUDINE VIATRIS contains
| Active ingredient (main ingredient) | lamivudine |
| Other ingredients (inactive ingredients) | Microcrystalline cellulose Sodium starch glycollate Type A Magnesium stearate Propylene glycol Opadry Complete Film Coating System 03H58736 White (ID 106640). |
Do not take LAMIVUDINE VIATRIS if you are allergic to any of these ingredients.
What LAMIVUDINE VIATRIS looks like
LAMIVUDINE VIATRIS 150 mg is a white to off-white film-coated, capsule shaped, biconvex tablet debossed with "M105" on one side of the tablet and a score line on the other side. (AUST R 167591).
LAMIVUDINE VIATRIS 300 mg is a white to off-white, film-coated, oval shaped, biconvex tablet debossed with "M300" on one side of the tablet and blank on the other side. (AUST R 167594).
LAMIVUDINE VIATRIS 150 mg is available in bottles of 60 tablets.
LAMIVUDINE VIATRIS 300 mg is available in bottles of 30 tablets.
Who distributes LAMIVUDINE VIATRIS
Alphapharm Pty Ltd trading as Viatris
Level 1, 30 The Bond
30-34 Hickson Road
Millers Point NSW 2000
www.viatris.com.au
Phone: 1800 274 276
This leaflet was prepared in December 2025.
LAMIVUDINE VIATRIS_cmi\Dec25/00
Brand Information
| Brand name | Lamivudine Viatris |
| Active ingredient | Lamivudine |
| Schedule | S4 |
MIMS Revision Date: 01 October 2023
1 Name of Medicine
Lamivudine.
2 Qualitative and Quantitative Composition
Lamivudine Viatris film-coated tablets contain 150 mg or 300 mg lamivudine as the active ingredient.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Lamivudine Viatris 150 mg film-coated tablets. White to off-white film-coated, capsule shaped, biconvex tablet debossed with "M105" on one side of the tablet and a score line on the other side.
Lamivudine Viatris 300 mg film-coated tablets. White to off-white, film-coated, oval shaped, biconvex tablet debossed with "M300" on one side of the tablet and blank on the other side.
4 Clinical Particulars
4.1 Therapeutic Indications
Lamivudine tablets in combination with other antiretroviral agents is indicated for the treatment of HIV infected adults and children.
4.2 Dose and Method of Administration
Food reduces the Cmax and extends the Tmax but the amount of drug absorbed is not reduced. The clinical significance of this is not known (see Section 5.2 Pharmacokinetic Properties).
Clinical studies indicate that lamivudine should be used in combination with other antiretroviral therapies. In controlled trials in adults, lamivudine was used in combination with zidovudine 200 mg tds (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Since accurate dosing cannot be achieved with this formulation, dosing according to weight bands is recommended for scored tablets. This dosing regimen for paediatric patients weighing 14-30 kg is based primarily on pharmacokinetic modelling. Therefore monitoring of efficacy and safety is necessary in these patients. More accurate dosing can be achieved with oral solution of lamivudine (see Section 5.2 Pharmacokinetic Properties, Pharmacokinetics in children).
To ensure administration of the entire recommended dose, the tablet(s) should be swallowed whole and not divided or crushed. If the patient is unable to swallow whole tablets, the tablets may be crushed and 100% of the crushed tablets could be added to a small amount of semisolid food or liquid, all of which should be consumed immediately (see Section 5.2 Pharmacokinetic Properties). Alternatively, lamivudine is available as an oral solution in another brand.
Adults, adolescents and children weighing at least 25 kg. The recommended dose of lamivudine is 150 mg twice daily. Alternatively, it may be administered as 300 mg once daily in patients who may benefit from a once daily regimen (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Children. Children less than three months of age. The limited data available are insufficient to propose specific dosage recommendations (see Section 5.1 Pharmacodynamic Properties, Clinical trials; Section 5.2 Pharmacokinetic Properties).
Children weighing 14 to < 20 kg. The recommended total daily dose of lamivudine is 150 mg. This may be administered as either one-half of a 150 mg scored tablet (75 mg) twice daily or one whole 150 mg tablet once daily.
Children weighing ≥ 20 kg to < 25 kg. The recommended total daily dose of lamivudine is 225 mg. This may be administered as either one-half of a 150 mg scored tablet (75 mg) in the morning and one whole 150 mg tablet in the evening, or one and a half 150 mg scored tablets (225 mg) once daily.
Children weighing at least 25 kg. The adult dosage of 150 mg twice daily or 300 mg once daily should be taken. Lamivudine is also available as an oral solution in another brand and lamivudine tablets should follow the dosing recommendations that are specific for the formulation (see Section 5.2 Pharmacokinetic Properties).
Data regarding the efficacy of once-daily dosing in paediatric population is limited to patients who transitioned from twice-daily to once-daily dosing after 36 weeks of treatment (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Renal impairment. Lamivudine plasma concentrations (AUC) are increased in patients with moderate to severe renal impairment due to decreased clearance. The dose should therefore be adjusted (see Tables 1 and 2). There are no data available on the use of lamivudine in children with renal impairment. The same percentage reduction in the adult dose is recommended for children with renal impairment. A reduction in the dose and/or an increase in the dosing interval should be considered in children aged at least three months and weighing less than 25 kg.
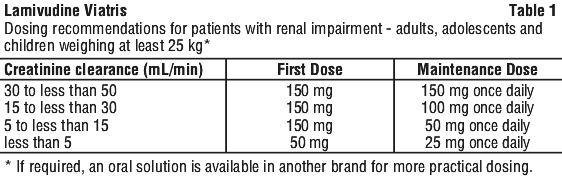

Hepatic impairment. Data obtained in patients with moderate to severe hepatic impairment shows that lamivudine pharmacokinetics are not significantly affected by hepatic dysfunction. Based on these data, no dose adjustment is necessary in patients with moderate or severe hepatic impairment unless accompanied by renal impairment.
4.3 Contraindications
The use of lamivudine is contraindicated in patients with known hypersensitivity to lamivudine or to any ingredient of the preparation.
4.4 Special Warnings and Precautions for Use
Lamivudine is not recommended for use as monotherapy.
Transmission of infection. While effective viral suppression with antiretroviral therapy has been proven to substantially reduce the risk of sexual transmission, a residual risk cannot be excluded. Precautions to prevent transmission should be taken in accordance with national guidelines.
Patients receiving lamivudine or any other antiretroviral therapy may continue to develop opportunistic infections and other complications of HIV infection, and therefore they should remain under close clinical observation by physicians experienced in the treatment of patients with associated HIV diseases.
Use with caution in the following circumstances. Pancreatitis. Pancreatitis has been observed in some patients receiving lamivudine. However it is unclear whether this was due to treatment with the medicinal product or to the underlying HIV disease. Pancreatitis must be considered whenever a patient develops abdominal pain, nausea, vomiting or elevated biochemical markers. Discontinue use of lamivudine until diagnosis of pancreatitis is excluded.
Pancreatitis in paediatric patients. In paediatric patients with a history of pancreatitis or other significant risk factors for the development of pancreatitis, lamivudine should be used with extreme caution and only if there is no satisfactory alternative therapy. Treatment with lamivudine should be stopped immediately if clinical signs, symptoms or laboratory abnormalities suggestive of pancreatitis occur (see Section 4.8 Adverse Effects (Undesirable Effects)).
Cirrhotic liver disease/hepatitis B virus. Clinical trial and marketed use of lamivudine, have shown that some patients with chronic hepatitis B virus (HBV) disease may experience clinical or laboratory evidence of recurrent hepatitis upon discontinuation of lamivudine, which may have more severe consequences in patients with decompensated liver disease. If lamivudine is discontinued in a patient with HIV and HBV coinfection, periodic monitoring of both liver function tests and markers of HBV replication should be considered.
Fat loss or fat gain. Fat loss or fat gain has been reported during combination antiretroviral therapy. The long term consequences of these events are currently unknown. A causal relationship has not been established.
Serum lipids and blood glucose. Serum lipid and blood glucose levels may increase during antiretroviral therapy. Disease control and lifestyle changes may also be contributing factors. Consideration should be given to the measurement of serum lipids and blood glucose. Lipid disorders should be managed as clinically appropriate.
Lactic acidosis and severe hepatomegaly with steatosis. Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of antiretroviral nucleoside analogues either alone or in combination, including lamivudine. A majority of these cases have been in women.
Clinical features which may be indicative of the development of lactic acidosis include generalised weakness, anorexia, and sudden unexplained weight loss, gastrointestinal symptoms and respiratory symptoms (dyspnoea and tachypnoea).
Caution should be exercised when administering lamivudine to any patient, and particularly to those with known risk factors for liver disease. Treatment with lamivudine should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis with or without hepatitis (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
Immune reconstitution syndrome. In HIV infected patients with severe immune deficiency at the time of initiation of antiretroviral therapy (ART), an inflammatory reaction to asymptomatic or residual opportunistic infections may arise and cause serious clinical conditions, or aggravation of symptoms. Typically, such reactions have been observed within the first few weeks or months of initiation of ART. Relevant examples are cytomegalovirus retinitis, generalised and/or focal mycobacterial infections and Pneumocystis jiroveci (P. carinii) pneumonia. Any inflammatory symptoms must be evaluated without delay and treatment initiated when necessary. Autoimmune disorders (such as Graves' disease, polymyositis and Guillain-Barre syndrome) have also been reported to occur in the setting of immune reconstitution, however the time to onset is more variable, and can occur many months after initiation of treatment and sometimes can be an atypical presentation.
Osteonecrosis. Although the etiology is considered to be multifactorial (including corticosteroid use, alcohol consumption, severe immunosuppression, higher body mass index), cases of osteonecrosis have been reported particularly in patients with advanced HIV-disease and/or long-term exposure to combination antiretroviral therapy. Patients should be advised to seek medical advice if they experience joint aches and pain, joint stiffness or difficulty in movement.
Use in hepatic impairment. See Section 4.2 Dose and Method of Administration, Hepatic impairment.
Use in renal impairment. In patients with moderate to severe renal impairment, the terminal plasma half-life of lamivudine exposure is increased due to decreased clearance. The dose should be adjusted (see Section 4.2 Dose and Method of Administration).
Use in the elderly. No data available.
Paediatric use. Children who at any time received lamivudine oral solution concomitantly with other antiretroviral oral solutions in clinical trials experienced lower rates of virological suppression, had lower plasma lamivudine exposure and developed viral resistance more frequently than children receiving tablets (see Section 5.1 Pharmacodynamic Properties, Clinical trials; Section 5.2 Pharmacokinetic Properties).
An all-tablet antiretroviral regimen should be used when possible. Lamivudine oral solution given concomitantly with sorbitol-containing medicines should be used only when the benefits of treatment outweigh possible risks including lower virological suppression. Consider more frequent monitoring of HIV-1 viral load when lamivudine is used with chronically-administered, sorbitol-containing medicines (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Effects on laboratory tests. See Section 4.8 Adverse Effects (Undesirable Effects).
4.5 Interactions with Other Medicines and Other Forms of Interactions
The likelihood of interactions is low due to limited metabolism and plasma protein binding and almost complete renal elimination of unchanged drug. Lamivudine is predominantly eliminated by active organic cationic secretion. The possibility of interactions with other drugs administered concurrently should be considered, particularly when their main route of elimination is active renal secretion via the organic cationic transport system e.g. trimethoprim. Other drugs (e.g. ranitidine, cimetidine) are eliminated only in part by this mechanism and were shown not to interact with lamivudine.
Coadministration of sorbitol solution (3.2 g, 10.2 g, 13.4 g) with a single 300 mg dose of lamivudine oral solution resulted in dose-dependent decreases (90% CI) of 14% (9-20%), 32% (28-37%), and 36% (32-41%) in lamivudine exposure (AUC∞) and 28% (20-34%), 52% (47-57%), and 55% (50-59%) in the Cmax of lamivudine in adults. When possible, avoid use of lamivudine with sorbitol-containing medicines or consider more frequent monitoring of HIV-1 viral load when chronic coadministration cannot be avoided (see Section 4.4 Special Warnings and Precautions for Use).
Effect of lamivudine on the pharmacokinetics of other agents. In vitro, lamivudine demonstrates no or weak inhibition of the drug transporters organic anion transporter 1B1 (OATP1B1), OATP1B3, breast cancer resistance protein (BCRP) or P-glycoprotein (Pgp), multidrug and toxin extrusion protein 1 (MATE1), MATE2-K or organic cation transporter 3 (OCT3). Lamivudine is therefore not expected to affect the plasma concentrations of drugs that are substrates of these drug transporters.
Lamivudine is an inhibitor of OCT1 and OCT2 in vitro with IC50 values of 17 and 33 microM, respectively, however lamivudine has low potential to affect the plasma concentrations of OCT1 and OCT2 substrates at therapeutic drug exposures (up to 300 mg).
Effect of other agents on the pharmacokinetics of lamivudine. Lamivudine is a substrate of MATE1, MATE2-K and OCT2 in vitro. Trimethoprim (an inhibitor of these drug transporters) has been shown to increase lamivudine plasma concentrations, however this interaction is not considered clinically significant as no dose adjustment of lamivudine is needed.
Lamivudine is a substrate of the hepatic uptake transporter OCT1. As hepatic elimination plays a minor role in the clearance of lamivudine, drug interactions due to inhibition of OCT1 are unlikely to be of clinical significance.
Lamivudine is a substrate of Pgp and BCRP, however due to its high bioavailability it is unlikely that these transporters play a significant role in the absorption of lamivudine. Therefore coadministration of drugs that are inhibitors of these efflux transporters is unlikely to affect the disposition and elimination of lamivudine.
Interactions relevant to lamivudine. Changes in zidovudine plasma levels when coadministered with lamivudine were not statistically significant. Zidovudine has no effect on the pharmacokinetics of lamivudine (see Section 5.2 Pharmacokinetic Properties).
Administration of trimethoprim, as trimethoprim/sulfamethoxazole 160 mg/800 mg increased lamivudine exposure by about 40%. However, unless the patient already has renal impairment, no dosage adjustment of lamivudine is necessary. Lamivudine has no effect on the pharmacokinetics of trimethoprim/sulfamethoxazole. Administration of lamivudine in patients with renal impairment should be assessed carefully. The effect of coadministration of lamivudine with higher doses of cotrimoxazole for the treatment of Pneumocystis carinii pneumonia and toxoplasmosis has not been studied.
In in vitro studies, ciprofloxacin, pentamidine and ganciclovir reduced the anti-HIV activity of lamivudine. The clinical significance of this is not known.
Lamivudine may inhibit the intracellular phosphorylation of zalcitabine when the two medicinal products are used concurrently. Lamivudine is therefore not recommended to be used in combination with zalcitabine.
Lamivudine may inhibit the intracellular phosphorylation of emtricitabine when the two medicinal products are used concurrently. Additionally, the mechanism of viral resistance for both lamivudine and emtricitabine is mediated via mutation of the same viral reverse transcriptase gene (M184V) and therefore the therapeutic efficacy of these drugs in combination therapy may be limited. Lamivudine is not recommended for use in combination with emtricitabine or emtricitabine-containing fixed dose combinations.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. No evidence of impaired fertility was seen in rats with exposures (based on Cmax) up to 70 times those observed at the clinical dosage.
Use in pregnancy. (Category B3)
Drugs which have been taken by only a limited number of pregnant women and women of childbearing age, without an increase in the frequency of malformation or other direct or indirect harmful effects on the human fetus having been observed. Studies in animals have shown evidence of an increased occurrence of fetal damage, the significance of which is considered uncertain in humans.
The Antiretroviral Pregnancy Registry has received reports of over 11,000 exposures to lamivudine during pregnancy resulting in live birth. These consist of over 4,200 exposures during the first trimester, over 6,900 exposures during the second/third trimester and included 135 and 198 birth defects respectively. The prevalence (95% CI) of defects in the first trimester was 3.2% (2.6, 3.7%) and in the second/third trimester, 2.8% (2.4, 3.2%). Among pregnant women in the reference population, the background rate of birth defects is 2.7%.
Studies in humans have confirmed that lamivudine crosses the placenta. Use in pregnancy should be considered only if the benefit outweighs the risk.
No evidence of teratogenicity was observed in rats and rabbits with exposure (based on Cmax) up to 40 and 36 times respectively those observed in humans at the clinical dosage. However, embryolethality was increased with consequent reduction in litter size in rabbits at exposures (based on both Cmax and AUC values) of less than those observed at the clinical dosage of 150 mg bd (approximately 4 mg/kg).
Lamivudine crossed the placenta in rats and rabbits. Although the results of animal studies are not always predictive of human response, the findings in the rabbit suggest a potential risk of early embryonic loss.
There have been reports of mild, transient elevations in serum lactate levels, which may be due to mitochondrial dysfunction, in neonates and infants exposed in utero or peri-partum to nucleoside reverse transcriptase inhibitors (NRTIs). The clinical relevance of transient elevations in serum lactate is unknown. There have also been very rare reports of developmental delay, seizures and other neurological disease. However, a causal relationship between these events and NRTI exposure in utero or peri-partum has not been established. These findings do not affect current recommendations to use antiretroviral therapy in pregnant women to prevent vertical transmission of HIV.
Use in lactation. Health experts recommend that where possible women infected with HIV do not breastfeed their infants in order to avoid the transmission of HIV. In settings where formula feeding is not feasible, local official lactation and treatment guidelines should be followed when considering breastfeeding during antiretroviral therapy.
A study in lactating rats showed that the concentration of lamivudine in milk was more than four times higher than that in maternal plasma. In a study following repeat oral dose of either 150 mg lamivudine twice daily (given in combination with 300 mg zidovudine twice daily) or 300 mg lamivudine twice daily, lamivudine was excreted in human breast milk (0.5 to 8.2 microgram/mL) at similar concentrations to those found in maternal serum.
In other studies following repeat oral dose of 150 mg lamivudine twice daily (given either in combination with 300 mg zidovudine or as lamivudine/zidovudine or abacavir/lamivudine/zidovudine) the breast milk:maternal plasma ratio ranged between 0.6 and 3.3. Lamivudine median infant serum concentrations ranged between 18 and 28 nanogram/mL and were not detectable in one of the studies (assay sensitivity 7 nanogram/mL). Intracellular lamivudine triphosphate (active metabolite of lamivudine) levels in the breastfed infants were not measured therefore the clinical relevance of the serum concentrations of the parent compound measured is unknown.
4.7 Effects on Ability to Drive and Use Machines
There have been no studies to investigate the effect of lamivudine on driving performance or the ability to operate machinery. Further, a detrimental effect on such activities cannot be predicted from the pharmacology of the drug. Nevertheless, the clinical status of the patient and the adverse event profile of both lamivudine and zidovudine should be borne in mind when considering the patient's ability to drive or operate machinery.
4.8 Adverse Effects (Undesirable Effects)
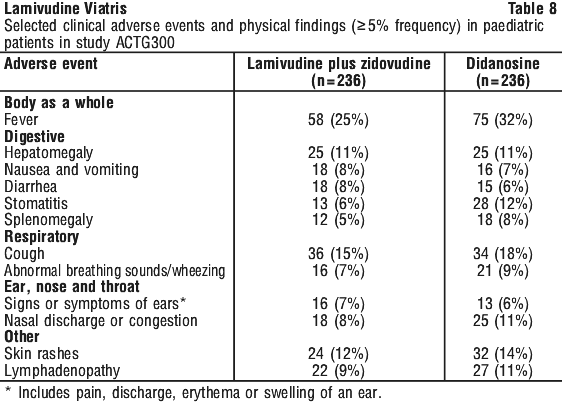
Table 3 lists all adverse events, occurring at an incidence of 5% or more, reported in controlled pivotal clinical trials in adults, irrespective of the investigator's assessment of the possible relationship to the study drug.
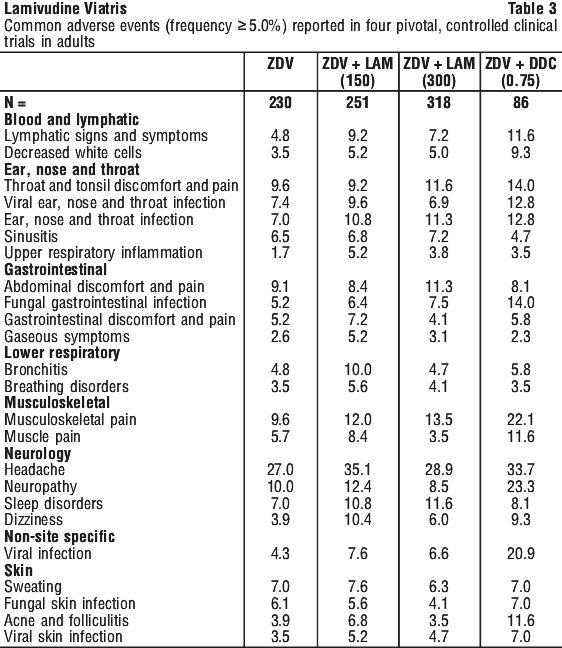
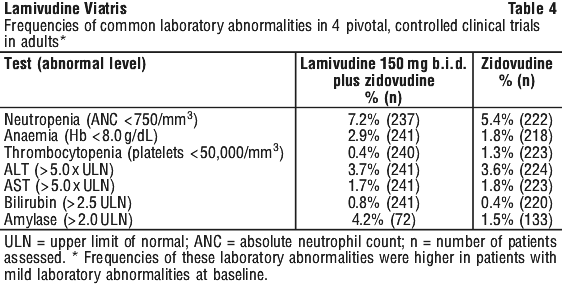
With many of these adverse reactions it is unclear whether they are drug related or are a result of the underlying disease.

In paediatric patients with a history of pancreatitis or other significant risk factors for the development of pancreatitis, the combination of lamivudine with other antiretroviral therapies should be used with extreme caution and only if there is no satisfactory alternative therapy.
Pancreatitis, which has been fatal in some cases has been observed in paediatric patients receiving lamivudine alone or in combination with other antiretroviral agents. In an open-label dose-escalation study, 14 patients (14%) developed pancreatitis while receiving monotherapy with lamivudine. Three of these patients died of complications of pancreatitis. In a second open label study, 12 patients (18%) developed pancreatitis. In both these open label studies, the paediatric subjects had advanced, symptomatic HIV infection and many had received extensive prior therapy. In addition, many of the children had predisposing medical conditions or medications which could have contributed to pancreatitis. In study ACTG300 in therapy-naïve subjects, pancreatitis was not observed in 236 patients randomised to lamivudine + zidovudine. Pancreatitis was observed in one patient in this study who received open-label lamivudine in combination with zidovudine and ritonavir following discontinuation of didanosine monotherapy. Paraesthesia and peripheral neuropathies were reported in 15 patients (15%) in one open study and 6 patients (9%) in the other open study and in 2 patients (1%) in ACTG300.
Table 6 lists all the adverse events, occurring at an incidence of 5% or more, reported in study EPV 20001.
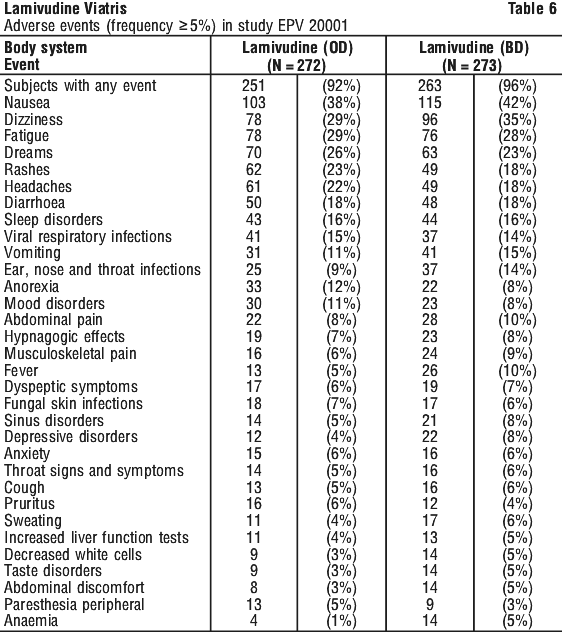

Adverse events and laboratory findings in study ACTG300 are provided in Tables 8 and 9.

The following convention has been utilised for the classification of undesirable effects: very common (> 1/10), common (> 1/100, < 1/10), uncommon (> 1/1,000, < 1/100), rare (> 1/10,000, < 1/1,000), very rare (< 1/10,000).
Musculoskeletal and connective tissue disorders. Common: arthralgia, muscle disorders. Rare: rhabdomyolysis.
Skin and subcutaneous tissue disorders. Common: rash, alopecia.
Blood and lymphatic systems disorders. Uncommon: neutropenia, anaemia, thrombocytopenia. Very rare: pure red cell aplasia.
Nervous system disorders. Common: headache. Very rare: paraesthesia. Peripheral neuropathy has been reported although a causal relationship to treatment is uncertain.
Gastrointestinal disorders. Common: nausea, vomiting, upper abdominal pain, diarrhoea. Rare: pancreatitis, although a causal relationship to treatment is uncertain. Rises in serum amylase.
Hepatobiliary disorders. Uncommon: transient rises in liver enzymes (AST, ALT).
Metabolism and nutrition disorders. Common: hyperlactataemia. Rare: lactic acidosis (see Section 4.4 Special Warnings and Precautions for Use).
General disorders and administrative site conditions. Common: fatigue, malaise, fever.
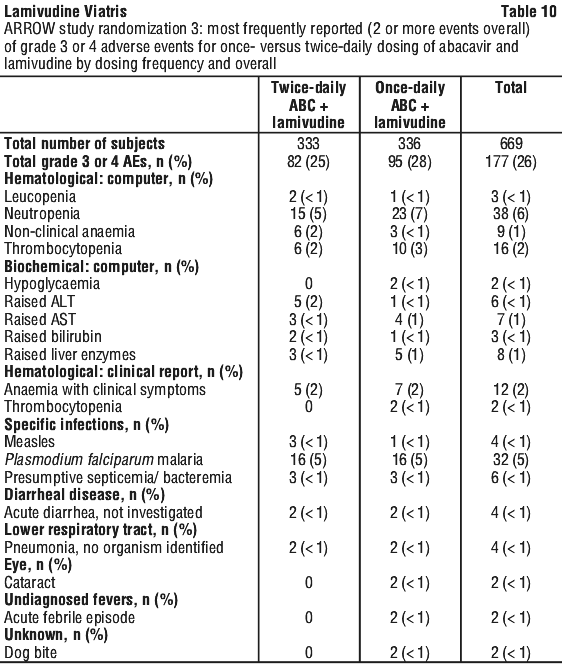
Paediatric population. The safety database to support lamivudine once daily dosing in paediatric patients comes from the ARROW Trial (COL105677) in which 669 HIV-1 infected paediatric subjects received abacavir and lamivudine either once or twice daily (see Section 5.1 Pharmacodynamic Properties, Clinical trials). Primary safety assessment in the ARROW trial was based on Grade 3 and Grade 4 adverse events. The frequency of Grade 3 and 4 adverse events was similar among subjects randomized to once-daily dosing compared with subjects randomized to twice-daily dosing (see Table 10). One event of Grade 4 hepatitis in the once-daily cohort was considered as uncertain causality by the investigator and all other Grade 3 or 4 adverse events were considered not related by the investigator.
4.9 Overdose
Administration of lamivudine at very high dose levels in acute animal studies did not result in any organ toxicity. No specific signs or symptoms have been identified following acute overdose with lamivudine, apart from those listed as undesirable effects. Since lamivudine is dialysable, continuous haemodialysis could be used in the treatment of overdosage, although this has not been studied.
Treatment of overdosage should be symptomatic and consist of general supportive measures.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action. Lamivudine is a potent, selective inhibitor of HIV-1 and HIV-2 replication in vitro. It is also active against zidovudine resistant clinical isolates of HIV.
In vitro, lamivudine demonstrates low cytotoxicity to peripheral blood lymphocytes, to established lymphocyte and monocyte-macrophage cell lines and to a variety of bone marrow progenitor cells. Lamivudine therefore has, in vitro, a high therapeutic index.
Lamivudine is metabolised intracellularly to the 5'-triphosphate which has an intracellular half-life of 10.5-15.5 hours. Lamivudine 5'-triphosphate is a weak inhibitor of the RNA and DNA dependent activities of HIV reverse transcriptase; its main mode of action is as a chain terminator of HIV reverse transcription.
Lamivudine does not interfere with cellular deoxynucleotide metabolism and has little effect on mammalian cell and mitochondrial DNA content.
The relationships between in vitro susceptibility of HIV to lamivudine and the clinical response to therapy remain under investigation. In vitro sensitivity testing has not been standardised and results may vary according to methodological factors.
Reduced in vitro sensitivity to lamivudine has been reported for HIV isolates from patients who have received lamivudine therapy.
No antagonistic effects in vitro were seen with lamivudine and other antiretrovirals (tested agents: abacavir, didanosine, nevirapine, zalcitabine and zidovudine).
In vitro studies show that restored sensitivity to zidovudine may occur following serial passage of zidovudine-resistant HIV-1 in increasing concentrations of lamivudine. Furthermore, in vivo, there is evidence showing that lamivudine plus zidovudine delays the emergence of zidovudine-resistant isolates in individuals with no prior antiretroviral therapy.
Drug resistance. Lamivudine resistant isolates of HIV-1 have been selected in vitro. The resistant isolates showed reduced susceptibility to lamivudine and genotypic analysis showed that the resistance was due to specific substitution mutations in the HIV-1 reverse transcriptase at codon 184 from methionine to either isoleucine or valine. HIV-1 strains resistant to both lamivudine and zidovudine have been isolated.
Susceptibility of clinical isolates to lamivudine and zidovudine was monitored in controlled clinical trials. In patients receiving lamivudine monotherapy or combination therapy with lamivudine plus zidovudine, HIV-1 isolates from most patients became phenotypically and genotypically resistant to lamivudine within 12 weeks. In some patients harbouring zidovudine-resistant virus at baseline, phenotypic sensitivity to zidovudine was restored by 12 weeks of treatment with lamivudine and zidovudine. Combination therapy with lamivudine plus zidovudine delayed the emergence of mutations conferring resistance to zidovudine.
Cross-resistance. Cross-resistance among certain reverse transcriptase inhibitors has been observed. Cross-resistance between lamivudine and zidovudine has not been reported. In some patients treated with lamivudine alone or in combination with zidovudine, isolates have emerged with a mutation at codon 184, which confers resistance to lamivudine. In the presence of the 184 mutation, cross-resistance to didanosine and zalcitabine has been seen in some patients; the clinical significance is unknown. In some patients treated with zidovudine plus didanosine or zalcitabine, isolates resistant to multiple reverse transcriptase inhibitors, including lamivudine, have emerged.
Clinical trial evidence from paediatric patients receiving lamivudine with other antiretroviral drugs (abacavir, nevirapine/efavirenz or zidovudine) has shown that the resistance profile observed in paediatric patients is similar to that observed in adults, in terms of the genotypic substitutions detected and their relative frequency.
Children receiving lamivudine oral solution concomitantly with other antiretroviral oral solutions in clinical trials developed viral resistance more frequently than children receiving tablets (see Section 4.4 Special Warnings and Precautions for Use; Section 5.1 Pharmacodynamic Properties, Clinical trials; Section 5.2 Pharmacokinetic Properties).
Clinical trials. Clinical endpoint study. Clinical end-point data from a prospective study indicate that lamivudine in combination with zidovudine alone or in combination with zidovudine containing treatment regimens results in a significant reduction in the risk of disease progression and mortality.
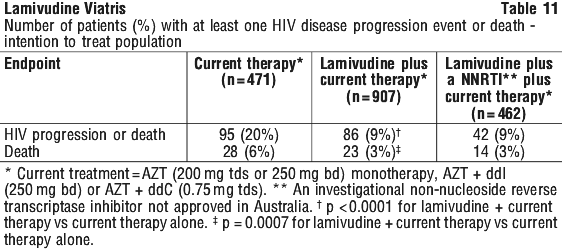
NUCB3007 (CAESAR) was a multicenter, double blind, placebo controlled study comparing continued current therapy [zidovudine (AZT) alone (62% of patients) or zidovudine with didanosine (ddI) or zalcitabine (ddC) (38% of patients)] to the addition of lamivudine or lamivudine plus an investigational non-nucleoside reverse transcriptase inhibitor, randomised 1:2:1. A total of 1,840 HIV infected adults with 25 to 250 (median, 126) CD4 cells/mm3 at baseline were enrolled: median age was 36 years, 87% were male, 83% were nucleoside experienced, and 17% were therapy naïve. The median duration of treatment for each group was current therapy* 327 days, lamivudine plus current therapy* 360 days and lamivudine plus NNRTI** plus current therapy* 360 days. Results are summarised in Table 11.
ACTG320 was a randomised, double-blind, placebo-controlled study to compare indinavir, zidovudine (or stavudine) and lamivudine with the 2 drug regimen of zidovudine (or stavudine) and lamivudine in HIV-infected patients with CD4 counts ≤ 200 cells/mm3. Patients had received ≥ 3 months prior zidovudine therapy and had no prior exposure to protease inhibitors. A total of 1156 patients were randomised. The median duration of follow-up was 38 weeks. During the study there were 96 new AIDS defining events or deaths, 63 (11%) in the zidovudine/lamivudine arm and 33 (6%) in the zidovudine/lamivudine/indinavir arm (estimated Hazard Ratio 0.50). There were 13 (6%) deaths in the zidovudine/lamivudine arm and 5 (2%) in the zidovudine/lamivudine/indinavir arm (Hazard Ratio 0.37). Both these results were statistically significant.
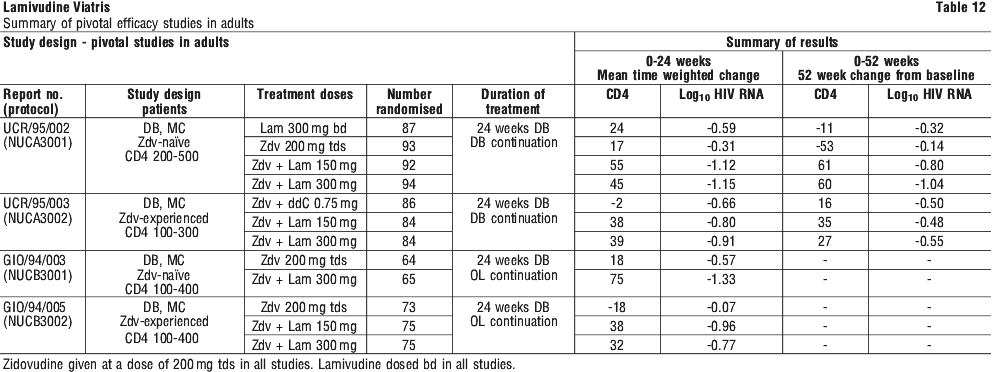
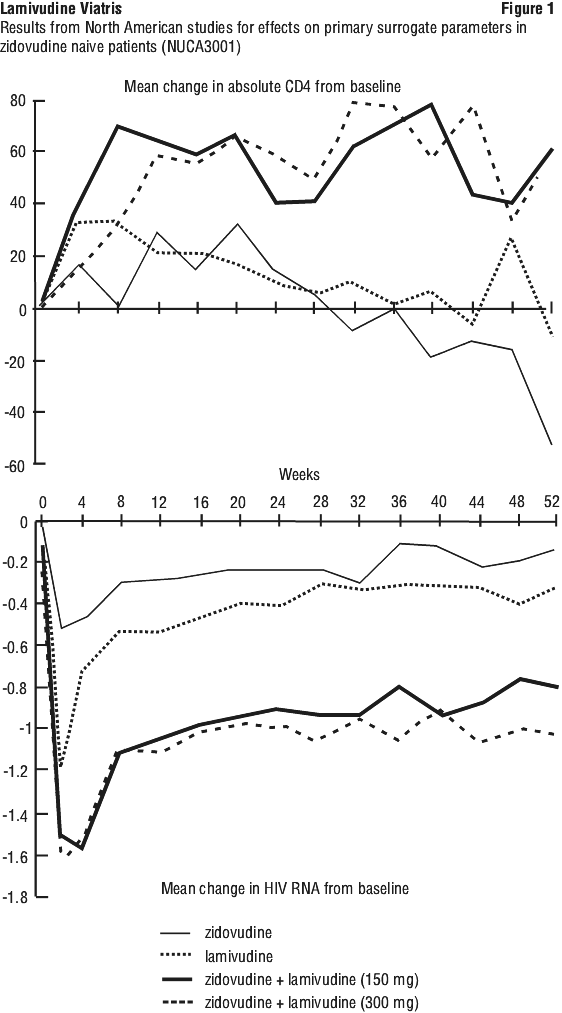
Surrogate endpoint studies in adults. Clinical efficacy in adults was based on the results of four pivotal studies in patients with or without prior antiretroviral therapy. Study designs are summarised in Table 12. All were randomised, double blind, multicentre studies. The characteristics of the patients at baseline are given in Table 13.
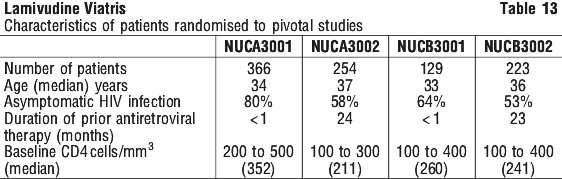
In the North American studies (NUCA3001 and NUCA3002) patients were allowed to remain in the study with blinding intact until the last patient had completed the 24 week assessment. Analysis of the subset of patients receiving treatment for at least 52 weeks established that the clinical benefits on CD4 cell count and viral load were maintained compared to zidovudine monotherapy over this period (p < 0.001). Results for CD4 count and log10 HIV RNA are given in Figures 1 and 2.
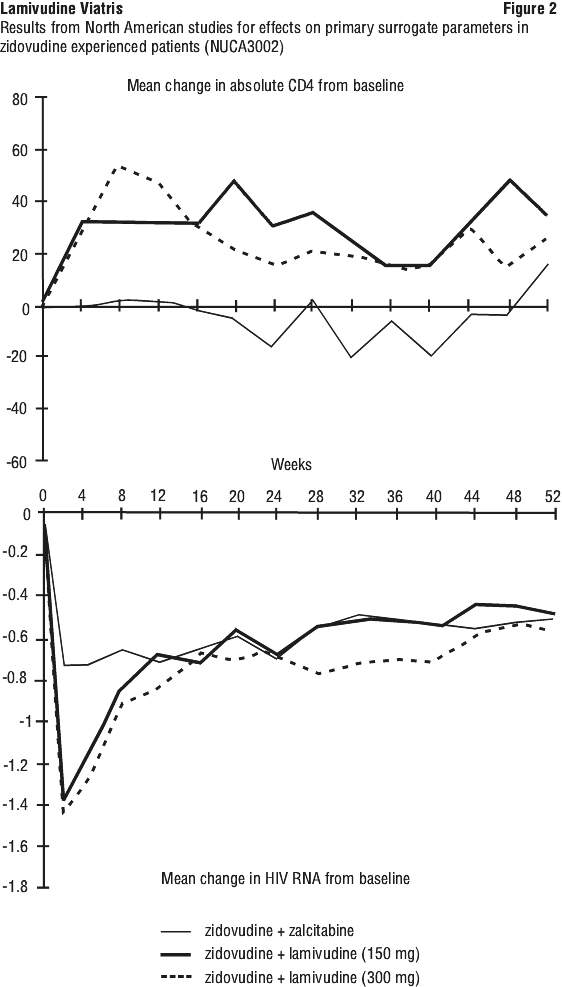
The proportions of subjects with plasma HIV-1 RNA < 400 copies/mL for intention-to-treat population using missing = failure analysis are summarised in Table 14.
Median CD4+ cell count values and changes from baseline are summarised by treatment group in Table 15.
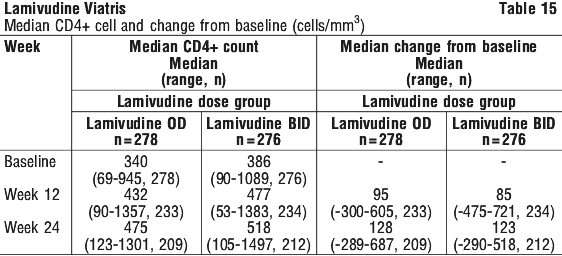
The results for Study EPV 20001 are reported to 24 weeks; this study is currently ongoing.
EPV40001 was a small, randomised, open label, controlled multicentre study in Thailand to evaluate the efficacy and safety of lamivudine once daily (OAD) versus lamivudine BID and abacavir (ABC) once daily versus ABC BID as components of a combination regimen including zidovudine (ZDV), lamivudine and ABC in antiretroviral-naïve, HIV-1 infected adults. Subjects were randomised to receive ZDV/lamivudine (BID)/ABC (BID) (control group), ZDV/lamivudine (OAD)/ABC (BID) or ZDV/lamivudine (BID)/ABC (OAD).
The three groups showed comparable efficacy using AAUCMB (Average Area Under Curve Minus Baseline) for HIV-1 RNA at week 48, the primary efficacy parameter for establishing noninferiority with a confidence interval of 0.4 log10 copies/mL (-2.0 log10 copies/mL control group, -2.0 log10 copies/mL lamivudine OAD group, -1.9 log10 copies/mL ABC OAD group) in the intent to treat exposed population (ITTE).
Using a secondary parameter of efficacy, the proportions of subjects with plasma HIV-1 RNA < 400 copies/mL for the ITTE population in a missing = failure analysis are summarised in Table 16:
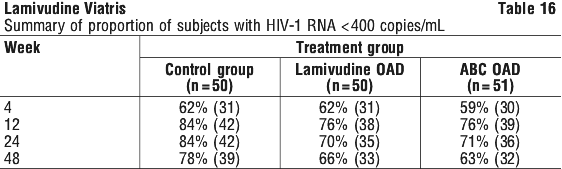
There were similar increases in median CD4+ cell counts observed in the three treatment groups at week 48 (control group: +216 cells/mm3; lamivudine OAD: +166 cells/mm3; ABC OAD: +152 cells/mm3).
Studies in children. Pharmacokinetic studies in children have established that relatively higher doses are required (8 mg/kg/day) to achieve comparable clinical exposure to that obtained with the recommended dose in adults (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties).
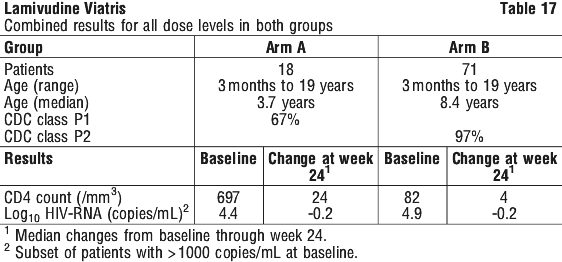
An open label, dose-escalation study (lamivudine monotherapy) was conducted in children aged 3 months to 17 years who had received no or minimal antiretroviral therapy (Arm A) or had experienced toxicity or become refractory to prior antiretroviral therapy (Arm B). At one centre compassionate treatment of patients with recurrent opportunistic infections was also allowed (Arm C). Patients were dosed at 1, 2, 4, 8, 12 or 20 mg/kg/day in two divided doses for 24 weeks. Dose escalation/reduction to 8 mg/kg/day was allowed after 24 weeks of treatment.
Lamivudine showed evidence of antiviral activity in both naive (Arm A) and experienced (Arm B) patients but no consistent dose response effects. The combined results for all dose levels in both groups are given in Table 17. No efficacy data are available on patients in Arm C.
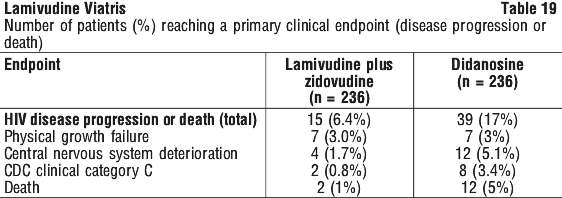
ACTG300 was a multicentre randomized double blind study, in paediatric patients, that provided for comparison of lamivudine plus zidovudine to didanosine monotherapy. The median duration on study medication was 10.1 months for patients receiving lamivudine + zidovudine and 9.2 months for children receiving didanosine monotherapy. Table 18 provides details of the changes from baseline for the two treatment groups and Table 19 the number of patients reaching primary clinical endpoint (disease progression or death):

Paediatric once daily dosing. A randomised comparison of a regimen including once daily vs twice daily dosing of abacavir and lamivudine was undertaken within a randomised, multicentre, controlled study of HIV infected, paediatric patients, conducted in Uganda and Zimbabwe. 1206 paediatric patients aged 3 months to 17 years enrolled in the ARROW Trial (COL105677) and were dosed according to the weight-band dosing recommendations in the World Health Organisation treatment guidelines (Antiretroviral therapy of HIV infection in infants and children, 2006). Subjects were ART naïve before enrolment and initiated treatment with an NNRTI + ABC (twice daily) + lamivudine (twice daily) with or without ZDV. After 36 weeks on antiretroviral therapy which included twice daily abacavir and lamivudine, 669 eligible children who had been on ART for at least 36 weeks, were currently taking lamivudine + abacavir twice daily as part of their ART regimen and expected to stay on these two drugs for at least the next 12 weeks were randomised to either continue twice daily dosing or switch to once daily abacavir and lamivudine for at least 96 weeks. At the time of the once daily versus twice daily randomisation, median age was 5.5 years (range 1.8-16.9 years). Most subjects (58.9%) were WHO stage 3 and most subjects (68.5%) had CD4 at ≥ 30%. The results are summarised in Table 20.
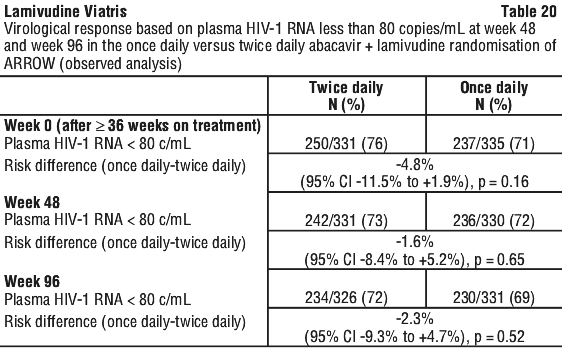
Note that the endpoint of viral load < 50 copies/mL could not be assessed due to low volumes of stored plasma samples from small children.
At the time of randomisation to once daily vs twice daily dosing (Week 0), those patients who had received tablet formulations had a higher rate of viral load suppression than those who had received any solution formulations at any time. These differences were observed in each different age group studied. This difference in suppression rates between tablets and solutions remained through Week 96 with once daily dosing. (See Table 21.)

5.2 Pharmacokinetic Properties
Absorption. Lamivudine is well absorbed from the gastrointestinal tract, and the bioavailability of oral lamivudine in adults is normally between 80 and 85%. Following oral administration the mean time (tmax) to maximal serum concentrations (Cmax) is about an hour. At therapeutic dose levels, i.e. approximately 4 mg/kg/day (as 150 mg twice daily) Cmax is in the order of 1-1.9 microgram/mL.
When a capsule formulation of lamivudine was ingested with food, there was a significant reduction in Cmax (47%) and extension to Tmax (2.2 hours). Although absorption of lamivudine was delayed, the amount of drug absorbed is not reduced.
The 150 mg tablet is bioequivalent and dose proportional to the 300 mg tablet with respect to AUC∞, Cmax and Tmax.
The plasma lamivudine half-life after oral dosing is 18 to 19 hours and the active moiety, intracellular lamivudine triphosphate, has a prolonged terminal half-life in the cell (16 to 19 hours). In 60 healthy adult volunteers, lamivudine 300 mg tablets once daily has been demonstrated to be pharmacokinetically equivalent at steady-state to lamivudine 150 mg tablets twice daily with respect to intracellular triphosphate AUC24 and Cmax.
The mean intracellular lamivudine triphosphate C0,ss was reduced by 16% and the mean C24,ss was reduced by 19% in volunteers given lamivudine 300 mg once daily compared with lamivudine 150 mg twice daily. The clinical relevance of the lower Cmin is unknown.
A study compared the pharmacokinetics of lamivudine tablets 300 mg once daily and lamivudine tablets 150 mg twice daily in 60 healthy, fasted volunteers. Steady-state plasma lamivudine AUCs were comparable for the two dosage regimens (mean ratio 0.94, 90% CI: 0.92-0.97) whereas the Cmax was increased by 66% (mean ratio 90% CI: 1.57-1.74) and Ctrough was 53% lower (mean ratio 90% CI: 0.44-0.50) with the 300 mg once daily. The clinical significance of the lower trough levels in unknown.
A bioequivalence study was conducted comparing the generic lamivudine 300 mg tablets with the originator lamivudine 300 mg tablets. The generic and originator mean Cmax for lamivudine was 2838.19 nanogram/mL and 3119.57 nanogram/mL, respectively. The Cmax point estimate for lamivudine was 0.9191 with the 90% confidence interval between 0.8424 and 1.0028. The mean AUC∞ point estimate for lamivudine was 0.9910 with the 90% confidence interval between 0.9215 and 1.0657. The Tmax for both the generic and originator tablets was 1.1 hours.
Administration of crushed tablets with a small amount of semi-food or liquid would not be expected to have an impact on the pharmaceutical quality, and would therefore not be expected to alter the clinical effect. This conclusion is based on the physiochemical and pharmacokinetic data, assuming that the patient crushes and transfers 100% of the tablet and ingests immediately.
Absorption differences have been observed between adult and paediatric populations (see Section 5.2 Pharmacokinetic Properties, Pharmacokinetics in children).
Distribution. From intravenous studies, the mean volume of distribution is 1.3 L/kg.
Limited data shows lamivudine penetrates the central nervous system and reaches the cerebrospinal fluid. The mean ratio CSF/serum lamivudine concentration 2-4 hours after oral administration was approximately 0.12. The true extent of penetration or relationship with any clinical efficacy is unknown.
Metabolism. Coadministration of zidovudine and lamivudine in a study of twelve asymptomatic patients with HIV caused minimal changes in lamivudine levels. The availability (mean AUC) of zidovudine was increased by 13%, mean Cmax was increased by 28%, and mean t1/2 reduced by 11%. There was considerable individual variation and the changes were not statistically significant. The relevance of these changes to clinical efficacy and safety is not known.
The likelihood of adverse drug interactions with lamivudine is low due to limited metabolism and plasma protein binding and almost complete renal elimination of unchanged drug. An interaction with trimethoprim, a constituent of trimethoprim with sulphamethoxazole causes a 40% increase in lamivudine exposure at therapeutic doses.
Excretion. The mean systemic clearance of lamivudine is approximately 0.32 L/kg/h, with predominantly renal clearance (> 70%) via active tubular secretion, but little (< 10%) hepatic metabolism.
A single dose pharmacokinetic study (n=16) in HIV-infected patients with normal renal function and with moderate (Clcr < 30 mL/min and > 10 mL/min) or end stage renal impairment (Clcr < 10 mL/min) showed there was a linear relationship between lamivudine clearance and renal function. A dosage adjustment is required in patients with creatinine clearance below 50 mL/min. Recommended dosage for patients with renal impairment is provided, see Section 4.2 Dose and Method of Administration.
A single dose pharmacokinetic study (n=24) in patients with moderate and severe hepatic impairment in comparison with healthy subjects showed no statistically significant differences for any of the mean pharmacokinetic parameters assessed. There was a trend towards reduced renal clearance in severely impaired subjects to an average of 76% (90% CI: 58% to 101%) relative to healthy control subjects.
Pharmacokinetics in children. The absolute bioavailability of lamivudine (approximately 58-66%) was lower and more variable in paediatric patients below 12 years of age. In children, administration of tablets given concomitantly with other antiretroviral tablets delivered higher plasma lamivudine AUC∞ and Cmax than oral solution given concomitantly with other antiretroviral oral solutions. Children receiving lamivudine oral solution according to the recommended dosage regimen achieve plasma lamivudine exposure within the range of values observed in adults. Children receiving lamivudine oral tablets according to the recommended dosage regimen achieve higher plasma lamivudine exposure than children receiving oral solution because higher mg/kg doses are administered with the tablet formulation and the tablet formulation has higher bioavailability (see Section 4.2 Dose and Method of Administration). Paediatric pharmacokinetic studies with both oral solution and tablet formulations have demonstrated that once daily dosing provides equivalent AUC0-24 to twice daily dosing of the same total daily dose. However, Cmax is approximately 2-fold higher with once daily dosing compared to twice daily dosing. (See Table 22.)

There are limited pharmacokinetic data for patients less than three months of age. In neonates one week of age, lamivudine oral clearance was reduced when compared to paediatric patients and is likely to be due to immature renal function and variable absorption. Therefore, to achieve similar adult and paediatric exposure, for neonates a dose of 2 mg/kg should be considered. Glomerular filtration estimates suggests that to achieve similar adult and paediatric exposure, the recommended dose for children aged six weeks and older could be 4 mg/kg twice a day. However there is no data available in neonates older than one week old.
5.3 Preclinical Safety Data
Genotoxicity. Lamivudine was not mutagenic in Salmonella typhimurium or E. coli reverse mutation assays with and without metabolic activation but did induce mutations at the thymidine kinase locus of the mouse lymphoma L5178Y cells without metabolic activation and was clastogenic in human peripheral blood lymphocytes, with and without metabolic activation in vitro. In rats lamivudine did not cause chromosomal damage in bone marrow cells in vivo or cause DNA damage in primary hepatocytes at estimated exposures many times higher than those observed clinically.
Lamivudine should not represent a genotoxic hazard to patients undergoing treatment.
Carcinogenicity. When lamivudine was administered orally to separate groups of rodents at doses up to 2000 times (mice and male rats) and 3000 (female rats) mg/kg/day, there was no evidence of a carcinogenic effect due to lamivudine in the mouse study. In the rat study there was an increased incidence of endometrial tumours at the highest dose (approximately 70 times the estimated human exposure at the recommended therapeutic dose of one tablet twice daily, based on AUC). However, the relationship of this increase to treatment is uncertain.
6 Pharmaceutical Particulars
6.1 List of Excipients
Microcrystalline cellulose, sodium starch glycollate Type A, magnesium stearate and the film coating contains propylene glycol with Opadry Complete Film Coating System 03H58736 White (ARTG PI No: 106640).
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C. Store in original container.
6.5 Nature and Contents of Container
Container type. HDPE bottle with PP child resistant closure.
Pack sizes. Lamivudine Viatris 150 mg. 60 tablets.
Lamivudine Viatris 300 mg. 30 tablets.
Some strengths, pack sizes and/or pack types may not be marketed.
Australian Register of Therapeutic Goods (ARTG). AUST R 167591. Lamivudine Viatris lamivudine 150 mg film-coated tablet bottle.
AUST R 167594. Lamivudine Viatris lamivudine 300 mg film-coated tablet bottle.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking it to your local pharmacy.
6.7 Physicochemical Properties
Lamivudine is a white to off white crystalline solid which is highly soluble in water.
Chemical structure. Chemical name: (2R-cis)-4-amino-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one.
Structural formula:
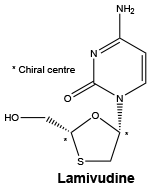
CAS number. 134678-17-4.
7 Medicine Schedule (Poisons Standard)
S4 (Prescription Only Medicine).
Date of First Approval
16 February 2012
Date of Revision
29 August 2023
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.