Lutathera
Brand Information
| Brand name | Lutathera |
| Active ingredient | Lutetium (177Lu) oxodotreotide |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Lutathera.
Summary CMI
LUTATHERA®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
▼ This medicine is new or being used differently. Please report side effects. See the full CMI for further details.
1. Why am I using LUTATHERA?
LUTATHERA contains the active ingredient lutetium (177Lu) oxodotreotide. LUTATHERA is used for the treatment of adults with certain tumours (gastroenteropancreatic neuroendocrine tumours). For more information, see Section 1. Why am I using LUTATHERA? in the full CMI.
2. What should I know before I use LUTATHERA?
Do not use if you have ever had an allergic reaction to lutetium (177Lu) oxodotreotide, amino acid solutions or any of the ingredients listed at the end of the CMI, you are pregnant or think you may be pregnant, if your kidneys are seriously impaired, or if you have any problem with the breakdown of amino acids in your body. The radiation coming from the medicine may potentially decrease your fertility. Talk to your doctor if you have any concerns, including whether you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use LUTATHERA? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with LUTATHERA and affect how it works. A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use LUTATHERA?
- LUTATHERA is given in a hospital or other licensed facility by specially trained staff.
- LUTATHERA is given as a single infusion into your vein approximately every 8 weeks for a total of 4 times.
More instructions can be found in Section 4. How do I use LUTATHERA? in the full CMI.
5. What should I know while using LUTATHERA?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using LUTATHERA? in the full CMI.
6. Are there any side effects?
Very common side effects include: feeling sick, vomiting, tiredness, tummy pain, diarrhoea. More serious very common side effects include: bleeding or bruising more easily than normal or difficulty to stop bleeding, infections with signs such as fever, sore throat or mouth ulcers, tiredness, weakness, pale skin, shortness of breath, passing less urine or smaller amounts than usual. For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
▼ This medicine is subject to additional monitoring. This will allow quick identification of new safety information. You can help by reporting any side effects you may get. You can report side effects to your doctor, or directly at www.tga.gov.au/reporting-problems.
LUTATHERA®
Active ingredient(s): Lutetium (177Lu) oxodotreotide
Consumer Medicine Information (CMI)
This leaflet provides important information about using LUTATHERA. You should also speak to your doctor, pharmacist or healthcare professional if you would like further information or if you have any concerns or questions about using LUTATHERA.
Where to find information in this leaflet:
1. Why am I using LUTATHERA?
2. What should I know before I use LUTATHERA?
3. What if I am taking other medicines?
4. How do I use LUTATHERA?
5. What should I know while using LUTATHERA?
6. Are there any side effects?
7. Product details
1. Why am I using LUTATHERA?
LUTATHERA contains the active ingredient lutetium (177Lu) oxodotreotide. LUTATHERA is a radiopharmaceutical product for therapy only.
LUTATHERA is used for the treatment of adults with certain tumors called gastroenteropancreatic neuroendocrine tumors. The tumour needs to have somatostatin proteins (receptors) on the surface of its cells in order for the medicine to work. LUTATHERA binds with these receptors and sends out radioactivity directly into the tumour cells, causing them to die.
The use of LUTATHERA involves exposure to amounts of radioactivity. Your doctor and the nuclear medicine doctor have considered that the clinical benefit that you will obtain from the procedure with the radiopharmaceutical outweighs the risk due to radiation.
2. What should I know before I use LUTATHERA?
Warnings
Do not use LUTATHERA if:
- You are allergic to lutetium (177Lu) oxodotreotide, or any of the ingredients listed at the end of this leaflet. Always check the ingredients to make sure you can use this medicine.
- You are pregnant or think you may be pregnant, or it has not been confirmed you are not pregnant. You need to make sure you are not pregnant by doing a pregnancy test before starting LUTATHERA.
- Your kidneys are seriously impaired.
- You are allergic to amino acid solutions or if you have any problem with the breakdown of amino acids in your body.
Check with your doctor if you:
- have or have had kidney problems
- have or have had liver problems
- have or have had any other type of cancer or previous anti-cancer treatment (chemotherapy) or radiation therapy
- have low levels of certain types of cells in the blood (red blood cells, white blood cells, neutrophils, and platelets)
- have high levels of potassium in your blood (symptoms can include breathlessness, weakness, numbness, chest pain, tremors or abnormal heart rhythm)
- have or have had heart failure
- take any medicines for any other condition.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
You must tell your doctor and/or the nuclear medicine doctor before taking LUTATHERA if there is a possibility you might be pregnant or if you have missed your period or if you are breast-feeding.
Do not take LUTATHERA if you are pregnant as ionising radiation is dangerous for the unborn baby. Stop breastfeeding during treatment with this medicine. If treatment with LUTATHERA during breastfeeding is necessary, the child must be weaned.
Contraception
For female patients, use effective birth control during LUTATHERA treatment and for 7 months after completing the treatment.
For male patients, use effective birth control during treatment and for 4 months after completing the treatment.
Ask your doctor, pharmacist or healthcare professional for options of effective birth control.
If you are a woman who could become pregnant, your doctor or other healthcare professional will check if you are pregnant and perform a pregnancy test, if necessary, before starting treatment with LUTATHERA.
If you become pregnant or think you are pregnant after starting treatment with LUTATHERA, tell your doctor and/or nuclear medicine doctor right away.
Fertility
The radiation coming from the medicine may potentially decrease your fertility. A consultation with a genetic counsellor is recommended if you wish to have children after treatment. Preservation of sperm or eggs may be offered to you before the treatment.
Children and Adolescents
LUTATHERA has not been studied in children and adolescents under 18 years of age.
You must limit extended close contact (less than one metre) with children to less than 15 minutes per day for 7 days after receiving each dose of LUTATHERA to minimise radiation risk.
Treatment monitoring
Your specialist or healthcare professional will do blood tests before and during treatment to check your condition and to notice any side effects as early as possible. Based on the results, your specialist may decide to delay, modify, or stop your treatment with LUTATHERA if necessary.
If necessary, the electrical activity of your heart will also be checked before you are discharged from the hospital (with a test called an electrocardiogram or ECG).
3. What if I am taking other medicines?
Tell your doctor, pharmacist or healthcare professional if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interfere with LUTATHERA and affect how it works. These include:
- somatostatin analog medicines that belong to a group of medicines called "anti-growth hormones" (somatostatin analogs) e.g. Sandostatin
- glucocorticoids (also called corticosteroids).
Check with your doctor, pharmacist or healthcare professional if you are not sure about what medicines, vitamins or supplements you are taking and if these affect LUTATHERA.
4. How do I use LUTATHERA?
How much will I receive
- The standard dose is 7400 MBq (megabecquerel, the unit used to express radioactivity) given directly into a vein.
When will I receive LUTATHERA
- LUTATHERA is given as a single infusion once approximately every 8 weeks for a total of 4 times.
Duration of treatment
- Your nuclear medicine doctor or other healthcare professional will tell you about the usual time required for the treatment.
- The infusion of LUTATHERA takes 30 ±10 minutes, but the complete time required will be approximately 5 hours. Your doctor will regularly monitor how you are going during this time.
During treatment with Lutathera
- During the procedure, you should isolate from other patients who are not receiving the same treatment due to the radiation released by this medicine.
- You will also be given an infusion with amino acids to protect your kidneys. This might make you feel sick and vomit and you will also receive an injection with a medicine before treatment to help decrease the sick feeling and vomiting.
- The doctor or other healthcare professional will inform you when you can leave the controlled area of the hospital.
If you miss an appointment to receive LUTATHERA
Contact your specialist or healthcare professional as soon as possible. You should receive LUTATHERA once every 8 weeks for a total of 4 times.
If you receive too much LUTATHERA
LUTATHERA is given in a hospital or other licensed facility. It is unlikely that you will receive too much. Your specialist will check and treat you if you receive too much.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using LUTATHERA?
Things you should do
- drink plenty of water e.g. 1 glass of water every hour so that you urinate as often as possible on the day of infusion and the day after, and try to empty your bowels every day, in order to remove the medicine from your body.
- limit close contact (less than 1 metre) with others in your household for 7 days after receiving each dose of LUTATHERA
- limit close contact (less than 1 metre) with children and pregnant women to less than 15 minutes per day for 7 days after receiving each dose of LUTATHERA
- sleep in a separate bedroom from others in your household for 7 days, from children or pregnant women for 15 days after receiving each dose of LUTATHERA.
Use of toilets
- Take special care to avoid contamination for 7 days after receiving treatment:
- You must always sit when using the toilet.
- It is important that you use toilet paper every time you use the toilet.
- Always wash your hands well after using the toilet.
- Flush all wipes and/or toilet paper down the toilet immediately after use.
- Flush any tissues or any other items that contain bodily waste, such as blood, urine, and faeces (poo) down the toilet. Items that cannot be flushed down the toilet, such as sanitary pads and bandages, must be placed in separate plastic waste disposal bags (according to "Household waste disposal" below).
Showering and laundry
- Take a shower every day for at least 7 days after treatment.
- Wash your underwear, pyjamas, sheets and any clothes that contain sweat, blood or urine separately from the laundry of other members of your household, using a standard washing cycle for 7 days after treatment. You do not need to use bleach and you do not need extra rinses.
For care givers
For 7 days after treatment:
- People who are confined to bed or have reduced mobility will preferably receive assistance from a care giver. The care giver must wear disposable gloves.
- Any special medical equipment that could be contaminated by your bodily fluids (e.g. catheters, colostomy bags, bedpans, water nozzles) must be emptied immediately into the toilet and then cleaned.
- Care givers who clean up vomit, blood, urine, or faeces (poo) should wear plastic gloves, which should be disposed of in a separate plastic waste disposal bag.
Household waste disposal
- Throw away all items in a separate plastic waste disposal bag that is to be used only for this purpose.
- Keep the plastic waste disposal bags separate from the other household waste and away from children and animals.
- A member of the hospital staff will tell you how and when to get rid of these waste disposal bags.
Hospitalisation and emergency care
- If for any reason you require emergency medical assistance or are unexpectedly admitted to the hospital during the 3 months after your treatment, please inform the healthcare professionals about the name, date, and dose of your radioactive treatment.
- Carry your discharge letter with you at all times to make it easier for 3 months after your treatment.
- Your specialist will inform you if you need to take any other special precautions after receiving this medicine. Contact your nuclear medicine doctor (your specialist) if you have any questions or are planning any travel.
Call your doctor straight away if you experience any of the following after the start of LUTATHERA treatment:
- flushing, diarrhoea, difficulty breathing with wheezing or coughing, dizziness, light-headedness (signs and symptoms of neuroendocrine hormone crisis), which may appear within the first 24 hours after LUTATHERA treatment.
- facial/throat swelling and/or difficulty breathing (signs and symptoms of angioedema).
- tiredness, loss of appetite, changes in your heartbeat, trouble thinking clearly (signs and symptoms of metabolic acidosis).
Remind any doctor, dentist, pharmacist or healthcare professional you visit that you are using LUTATHERA.
Things you should not do
- Do not stop using effective birth control during LUTATHERA treatment and for 7 months after completing the treatment for females and for 4 months after completing the treatment for males.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how LUTATHERA affects you.
LUTATHERA may cause dizziness and tiredness in some people.
Drinking alcohol
Tell your doctor if you drink alcohol.
Looking after your medicine
- You will not have to store this medicine. This medicine is stored under the responsibility of the specialist where you were treated. Storage of radiopharmaceuticals will be in accordance with Australian regulations on radioactive materials.
- The storage instructions are for the specialist. They are: Store below 25°C. Do not freeze LUTATHERA. Store in the original package to protect from ionising radiation (lead shielding).
Getting rid of any unwanted medicine
LUTATHERA is only given in special facilities by appropriately qualified staff. They will be required to dispose of the medicine following treatment in accordance with Australian laws.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor, pharmacist or healthcare professional if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
Tummy problems:
| Speak to your doctor if you have any of these less serious side effects and they worry you. If these side effects become severe, please tell your doctor, or specialist |
Serious side effects
| Serious side effects | What to do |
Blood problems:
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor, pharmacist or healthcare professional if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
What LUTATHERA contains
| Active ingredient (main ingredient) | lutetium (177Lu) oxodotreotide |
| Other ingredients (inactive ingredients) | Acetic acid Sodium acetate Gentisic acid Ascorbic acid Pentetic acid Sodium chloride Sodium hydroxide Water for injections |
Do not use this medicine if you are allergic to any of these ingredients.
What LUTATHERA looks like
LUTATHERA is supplied in a clear, colourless type I glass vial, closed with a bromobutyl rubber stopper and aluminium cap.
Each vial contains a volume that ranges from 20.5 to 25.0 mL of solution, corresponding to a radioactivity of 7400 MBq ± 10% at the date and time of infusion.
The vial is enclosed within a lead shielded container and placed in a plastic sealed container.
(Aust R 455452).
Who distributes LUTATHERA
LUTATHERA is supplied in Australia by:
Novartis Pharmaceuticals Australia Pty Limited
ABN 18 004 244 160
54 Waterloo Road
Macquarie Park NSW 2113
Telephone 1 800 671 203
Website: www.novartis.com.au
This leaflet was prepared in November 2025.
Registered trademark of Novartis.
© Copyright Novartis 2025
Internal document code: (lut141125c) based on PI (lut141125i)
Brand Information
| Brand name | Lutathera |
| Active ingredient | Lutetium (177Lu) oxodotreotide |
| Schedule | S4 |
▼ This medicinal product is subject to additional monitoring in Australia. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse events at www.tga.gov.au/reporting-problems.
MIMS Revision Date: 01 February 2026
1 Name of Medicine
Lutetium (177Lu) oxodotreotide.
2 Qualitative and Quantitative Composition
For the full list of excipients, see Section 6.1 List of Excipients.
One mL of solution contains 370 MBq of lutetium (177Lu) oxodotreotide at the date and time of calibration.
The total amount of radioactivity per single-dose vial is 7400 MBq ± 10% at the date and time of infusion. Given the fixed volumetric activity of 370 MBq/mL at the date and time of calibration, the volume of the solution in the vial ranges between 20.5 and 25.0 mL to provide the required amount of radioactivity at the date and time of infusion.
3 Pharmaceutical Form
Solution for intravenous infusion. Lutathera is a clear, colourless to slightly yellow solution with a pH range of 4.5 to 6.0.
4 Clinical Particulars
4.1 Therapeutic Indications
Lutathera is indicated for the treatment of somatostatin receptor-positive gastroenteropancreatic neuroendocrine tumours (GEP-NETs), including foregut, midgut, and hindgut neuroendocrine tumours in adults.
4.2 Dose and Method of Administration
Important safety instructions. Lutathera is a radiopharmaceutical and should be handled with appropriate safety measures to minimise radiation exposure in accordance with national regulations and/or institutional guidelines (see Section 4.4 Special Warnings and Precautions for Use). Waterproof gloves and effective radiation shielding should be used when handling Lutathera (see Section 6.6 Special Precautions for Disposal).
Radiopharmaceuticals, including Lutathera, should be used by or under the control of physicians who are qualified by specific training and experienced in the safe use and handling of radiopharmaceuticals, and whose experience and training have been approved by the appropriate governmental agency authorised to license the use of radiopharmaceuticals.
Pregnancy status of females of reproductive potential must be verified prior to initiating treatment with Lutathera (see Section 4.6 Fertility, Pregnancy and Lactation).
Monitor patients closely for signs and symptoms of hypersensitivity reactions during and following Lutathera administration for a minimum of 2 hours (see Section 4.4 Special Warnings and Precautions for Use).
Patient identification. Before initiating treatment with Lutathera, presence of somatostatin receptor-positive tumours must be confirmed, preferably by somatostatin receptor imaging.
Dosage regimen. General target population. Administer premedications and concomitant medications as recommended.
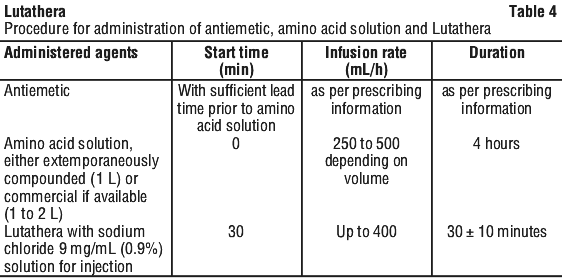
Adults. The recommended treatment regimen of Lutathera in adults consists of 4 infusions of 7400 MBq each. The recommended interval between each infusion is 8 weeks (± 1 week).
Antiemetics. Antiemetics should be administered with sufficient lead time prior to the start of the amino acid solution. Refer to full product information of antiemetics for administration instructions.
In case of severe nausea or vomiting during the infusion of the amino acid solution despite administration of a pre-procedure antiemetic, an antiemetic of a different pharmacological class can be administered (see Section 4.4 Special Warnings and Precautions for Use; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
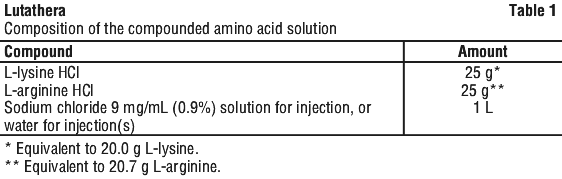
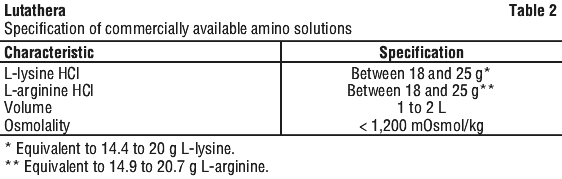
Amino acid solution. For renal protection, an intravenous amino acid solution containing L-lysine and L-arginine must be administered 30 minutes before the start of the Lutathera infusion (see Tables 1 and 2). The amino acid solution infusion should continue during and for at least 3 hours after the completion of the Lutathera infusion. Infusion of the amino acid solution and Lutathera through a separate venous access in each of the patient's arms is the preferred method. However, if two intravenous lines are not possible due to poor venous access or institutional/clinical preference, the amino acid solution and Lutathera may be infused through the same line via a three-way valve, taking into consideration flow rate and maintenance of venous access. The dose of the amino acid solution should not be decreased even if a reduced dose of Lutathera is administered.
An amino acid solution containing just L-lysine and L-arginine in the amounts specified in Table 1 is considered the medicinal product of choice, due to the lower total volume to be infused and lower osmolality.
The amino acid solution can be prepared as a compounded product, in compliance with the hospital's good preparation practices for sterile medicinal products and according to the composition specified in Table 1.
These laboratory tests should be performed shortly before each administration and between 4 to 6 weeks after each dose of Lutathera. It is also recommended to perform these tests every 4 weeks for at least 3 months after the last infusion of Lutathera and every 6 months thereafter, in order to be able to detect possible delayed adverse drug reactions (ADRs) (see Section 4.8 Adverse Effects (Undesirable Effects)). Dosing may need to be modified based on the test results as described in Table 3 Recommended dose modifications for adverse drug reactions.
Treatment monitoring (administration of amino acid solution). Test serum potassium levels before each administration of amino acid solution (see Section 4.4 Special Warnings and Precautions for Use, Hyperkalaemia).
Dose modifications for adverse drug reactions. Management of severe or intolerable adverse drug reactions may require temporary dose interruption (extension of the dosing interval from 8 weeks up to 16 weeks), dose reduction, or permanent discontinuation of treatment with Lutathera. Recommended dose modifications of Lutathera for adverse drug reactions are provided in Table 3.

intercurrent disease (e.g. urinary tract infection) which the physician considers could increase the risks associated with Lutathera administration. Resume treatment when resolved or stabilised;
major surgery (withhold Lutathera for 12 weeks after the date of surgery).
Special populations. Use in renal impairment. Lutathera is substantially excreted by the kidneys, thus patients with renal impairment may be at increased risk of toxicity due to increased radiation exposure. The pharmacokinetic profile and safety of Lutathera in patients with baseline severe renal impairment (creatinine clearance < 30 mL/min by Cockcroft-Gault formula) or end-stage renal disease have not been studied. Treatment with Lutathera in patients with kidney failure with creatinine clearance < 30 mL/min is contraindicated. Treatment with Lutathera in patients with baseline creatinine clearance < 40 mL/min is not recommended. No dose adjustment is recommended for renally impaired patients with baseline creatinine clearance ≥ 40 mL/min. However, renal function should be monitored more frequently during treatment as these patients may be at greater risk of toxicity.
Patients with a history of renal impairment and high tumour burden may be at greater risk of tumour lysis syndrome and should be treated with increased caution.
Use in hepatic impairment. Patients with hepatic impairment may be at increased risk of hepatotoxicity due to radiation exposure. The pharmacokinetic profile and safety of Lutathera in patients with baseline severe hepatic impairment (total bilirubin > 3 times upper limit of normal, regardless of AST level) have not been studied. Patients with baseline hepatic impairment with either total bilirubin > 3 times the upper limit of normal or albuminemia < 30 g/L and INR > 1.5 should only be treated with Lutathera after a careful benefit-risk assessment. No dose adjustment is recommended for patients with baseline mild or moderate hepatic impairment.
Paediatric use (below 18 years). The safety and efficacy of Lutathera have not been established in paediatric patients.
Use in the elderly (65 years of age or above). No dosage adjustment is required in patients 65 years of age or above as clinical experience has not identified differences in responses between geriatric and younger patients. However, close follow-up allowing for prompt dose adaptation in this population is advisable.
Method of administration. Lutathera is a ready-to-use solution for intravenous infusion for single use in one patient only. Discard any unused medicinal product.
Preparation instructions. Use aseptic technique and radiation shielding when administering the Lutathera solution. Use tongs when handling the vial to minimise radiation exposure.
Visually inspect the product under a shielded screen for particulate matter and discolouration prior to administration. Discard the vial if particulates and/or discolouration are present.
Inspect the package for damage and use a calibrated radioactivity measurement system to determine if any radioactive contamination is present. Do not use the product if the integrity of the vial or the lead container is compromised.
Do not inject the Lutathera solution directly into any other intravenous solution.
Confirm the amount of radioactivity of Lutathera delivered to the patient with a calibrated radioactivity measurement system prior to and after each Lutathera administration to confirm that the actual amount of radioactivity administered is equal to the planned amount.
Do not administer Lutathera as an intravenous bolus.
Soon after the start of the infusion, monitor the radioactivity emission from the patient using a calibrated radioactivity measurement system to ensure the dose is delivered. During the infusion, the radioactivity emission from the patient should steadily increase, while that from the Lutathera vial should decrease.
Careful monitoring of the patient's vital signs during the infusion is recommended.
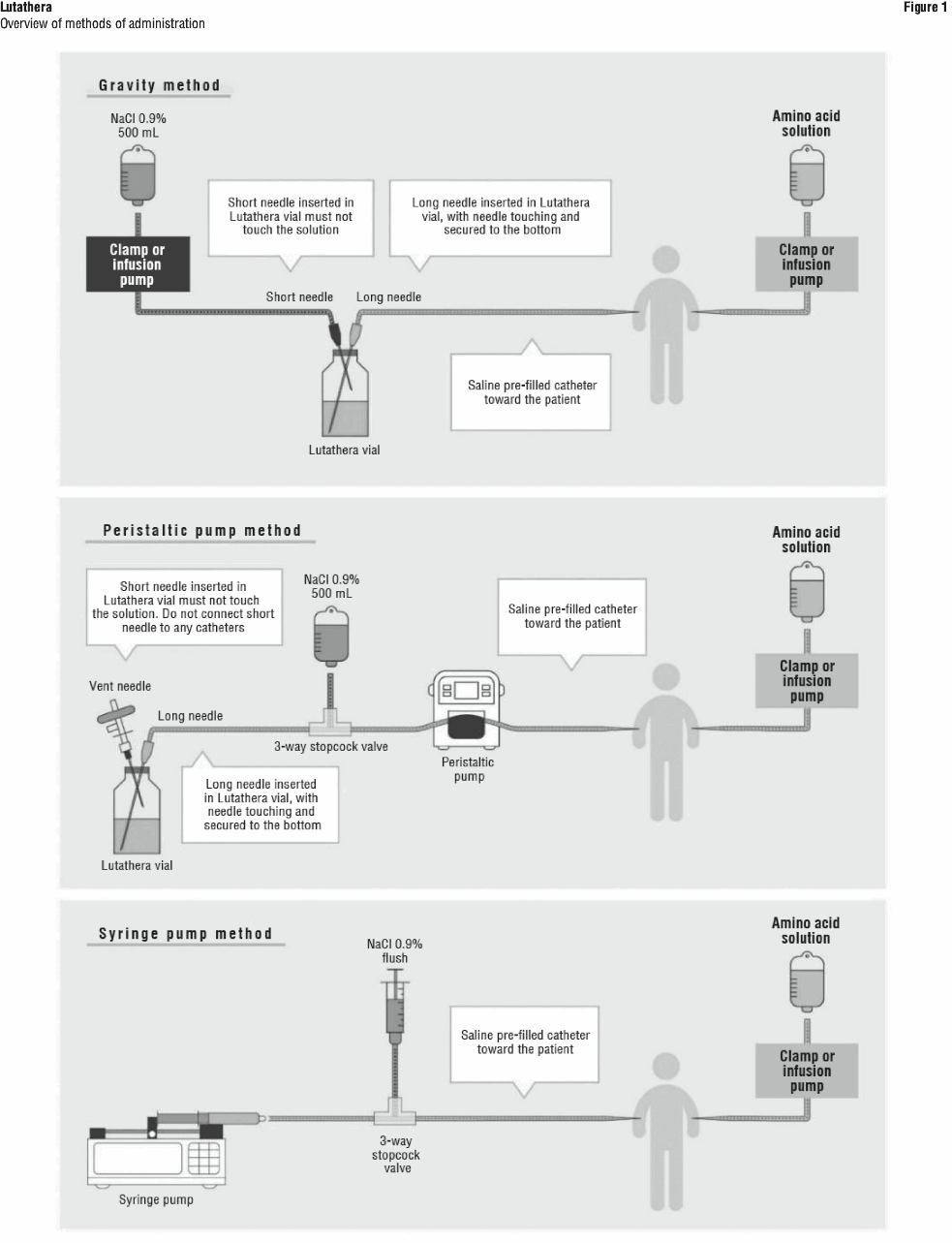
Administration instructions. The gravity method, the peristaltic pump method or the syringe pump method may be used for administration of the recommended dose. Treating healthcare professionals may use other methods deemed appropriate and safe, particularly when dose reduction is required. When using the gravity method or the peristaltic pump method, Lutathera should be infused directly from its original container. The peristaltic pump method or the syringe pump method should be used when administering a reduced dose of Lutathera following a dose modification for an adverse drug reaction (see Table 3 Recommended dose modifications for adverse drug reactions). Using the gravity method to administer a reduced dose of Lutathera may result in the delivery of the incorrect volume of Lutathera if the dose is not adjusted prior to administration. Radiation safety precautions must be considered regardless of the administration method used (see Section 4.4 Special Warnings and Precautions for Use).
Table 4 summarises the whole administration procedure for Lutathera:
Insert a second needle that is 9 cm, 18-gauge (long needle) into the Lutathera vial, ensuring that this long needle touches and is secured to the bottom of the Lutathera vial during the entire infusion. Connect the long needle to the patient by an intravenous catheter that is pre-filled with 0.9% sterile sodium chloride solution and that is used for the Lutathera infusion into the patient.
Use a clamp or an infusion pump to regulate the flow of the sodium chloride solution via the short needle into the Lutathera vial. The sodium chloride solution entering the vial through the short needle will carry the Lutathera solution from the vial to the patient via the intravenous catheter connected to the long needle over a total duration of 30 ± 10 minutes, at an infusion rate of up to 400 mL/h. The infusion should start at a lower rate of < 100 mL/h for the first 5 to 10 minutes and should then be increased depending on the patient's venous status. Constant intra vial pressure should be maintained during the entire infusion.
During the infusion, ensure that the level of solution in the Lutathera vial remains constant by repeated direct visual control when transparent shielded container is used or using a pair of tongs to handle the vial when the lead shipping container is used.
Monitor the flow of Lutathera from the vial to the patient during the entire infusion.
Disconnect the vial from the long needle line and clamp the saline line once the level of radioactivity is stable for at least five minutes.
Follow the infusion with an intravenous flush of 25 mL of 0.9% sterile sodium chloride solution through the intravenous catheter to the patient.
Instructions for the peristaltic pump method. Insert a filtered 2.5 cm, 20-gauge needle (short venting needle) into the Lutathera vial. Ensure that the short needle does not touch the Lutathera solution in the vial and do not connect the short needle directly to the patient or to the peristaltic pump.
Insert a second needle that is 9 cm, 18-gauge (long needle) into the Lutathera vial, ensuring that the long needle touches and is secured to the bottom of the Lutathera vial during the entire infusion. Connect the long needle and a 0.9% sterile sodium chloride solution to a 3-way stopcock valve via appropriate tubing.
Connect the output of the 3-way stopcock valve to tubing installed on the input side of the peristaltic pump following the pump manufacturer's instructions.
Prime the line by opening the 3-way stopcock valve and pumping the Lutathera solution through the tubing until it reaches the exit of the valve.
Prime the intravenous catheter which will be connected to the patient by opening the 3-way stopcock valve to the 0.9% sterile sodium chloride solution and pumping the 0.9% sterile sodium chloride solution until it exits the end of the catheter tubing.
Connect the primed intravenous catheter to the patient and set the 3-way stopcock valve such that the Lutathera solution is in line with the peristaltic pump.
Infuse an appropriate volume of Lutathera solution over a 30 ± 10 minute period to deliver the desired radioactivity.
When the desired Lutathera radioactivity has been delivered, stop the peristaltic pump, and then change the position of the 3-way stopcock valve so that the peristaltic pump is in line with the 0.9% sterile sodium chloride solution. Restart the peristaltic pump and infuse an intravenous flush of 25 mL of 0.9% sterile sodium chloride solution through the intravenous catheter to the patient.
Instructions for the syringe pump method. Withdraw an appropriate volume of Lutathera solution to deliver the desired radioactivity by using a disposable syringe fitted with a syringe shield and a disposable sterile needle that is 9 cm, 18-gauge (long needle). To aid the withdrawal of the solution, it is possible to use a filtered 2.5 cm, 20-gauge needle (short venting needle) to reduce the resistance from the pressurised vial. Ensure that the short needle does not touch the Lutathera solution in the vial.
Fit the syringe into the shielded pump and include a 3-way stopcock valve between the syringe and an intravenous catheter that is pre-filled with 0.9% sterile sodium chloride solution and that is used for Lutathera administration to the patient.
Infuse an appropriate volume of Lutathera solution over a 30 ± 10 minute period to deliver the desired radioactivity.
When the desired Lutathera radioactivity has been delivered, stop the syringe pump, and then change the position of the 3-way stopcock valve so to flush the syringe with 25 mL of 0.9% sterile sodium chloride solution. Restart the syringe pump.
After the flush of the syringe has been completed, perform an intravenous flush with 25 mL of 0.9% sterile sodium chloride solution through the intravenous catheter to the patient.
See Figure 1.
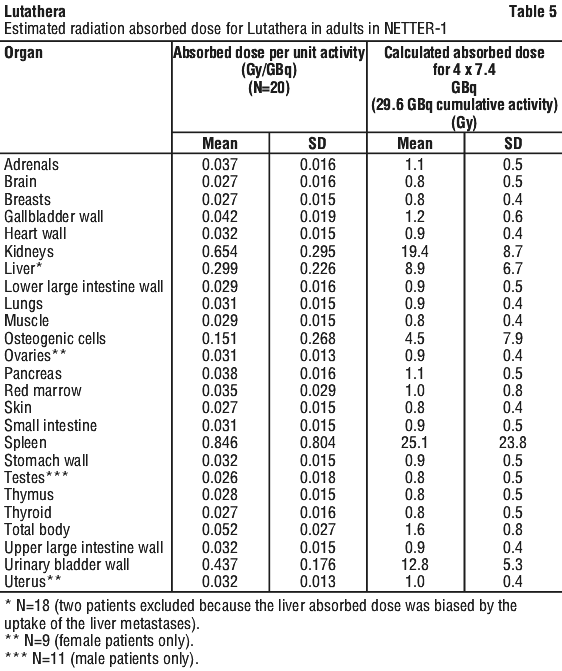
The mean and standard deviation (SD) of the estimated radiation absorbed doses for adults receiving Lutathera are shown in Table 5.
4.3 Contraindications
Established or suspected pregnancy or when pregnancy has not been excluded (see Section 4.6 Fertility, Pregnancy and Lactation).
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of Excipients.
The use of Lutathera is contraindicated in patients with kidney failure with creatinine clearance < 30 mL/min.
Hypersensitivity to one or more amino acids or congenital abnormality of amino acid metabolism.
4.4 Special Warnings and Precautions for Use
Risk from radiation exposure. Lutathera contributes to a patient's overall long-term cumulative radiation exposure. Exposure to ionising radiation is linked with cancer induction and a potential for development of hereditary defects. The radiation dose resulting from therapeutic exposure may result in higher incidence of cancer and mutations. Patients under treatment with Lutathera should be kept away from others during administration and until the radiation emission limits stipulated by the applicable laws are reached, usually within the 4 to 5 hours following Lutathera administration (see Section 5.2 Pharmacokinetic Properties). Radiation exposure should be minimised to patients, medical personnel, and household contacts during and after treatment with Lutathera for at least 7 days (see Section 4.4 Special Warnings and Precautions for Use, Radioprotection rules) and also consistent with institutional good radiation safety practices and patient management procedures (see Section 4.2 Dose and Method of Administration).
Myelosuppression. Myelosuppression was reported in the majority of patients treated with Lutathera (see Section 4.8 Adverse Effects (Undesirable Effects)).
Most of the haematological events were mild or moderate and reversible/transient. Patients with impaired bone marrow function, as well as patients who received prior chemotherapy or external beam radiotherapy (involving more than 25% of the bone marrow) may be at higher risk of haematological toxicity during Lutathera treatment.
Haematological evaluation of patients must be performed at baseline and prior to each dose of Lutathera. Based on the severity of myelosuppression, Lutathera may require withholding, dose reduction, or permanent discontinuation as described in Table 3 Recommended dose modifications for adverse drug reactions.
Treatment of patients with severely impaired haematological function at baseline and during treatment (e.g. Hb < 4.9 mmol/L or 8 g/dL, platelets < 75 x 109/L or leukocytes < 2 x 109/L) is not recommended unless solely due to lymphopenia.
Secondary myelodysplastic syndrome and acute leukemia. Late-onset myelodysplastic syndrome (MDS) and acute leukemia (AL) have been reported after treatment with Lutathera (see Section 4.8 Adverse Effects (Undesirable Effects)).
In a phase III study (NETTER-1), with a median follow-up time of 76 months in the main study, MDS was reported in 3 patients (2 patients from the main study and 1 patient from the dosimetry sub-study) (2.3%) receiving Lutathera compared to no patients who received high-dose octreotide LAR.
In a phase I/II study (ERASMUS), 16 patients (2%) developed MDS and 4 (0.5%) developed AL. The median time to onset was 29 months (9 to 45 months) for MDS and 55 months (32 to 125 months) for AL.
Renal toxicity. Renal dysfunction can develop during and after treatment with Lutathera. Cases of chronic renal impairment have been reported in patients several years following treatment with Lutathera which were mild in nature and were confirmed by serum/urine analyses (see Section 4.8 Adverse Effects (Undesirable Effects)).
In ERASMUS, 8 patients (1%) developed renal failure 3 to 36 months following treatment with Lutathera.
Administration of amino acid solution should start 30 minutes before and should continue during and for at least 3 hours after each Lutathera dose (see Section 4.2 Dose and Method of Administration). The amino acid solution helps to decrease reabsorption of lutetium (177Lu) oxodotreotide through the proximal tubules resulting in decrease in the radiation dose to the kidneys. Patients should be encouraged to remain hydrated and to urinate frequently before, on the day of and the day after administration of Lutathera (e.g. 1 glass of water every hour).
Renal function as determined by serum creatinine and creatinine clearance as calculated using Cockcroft-Gault formula or an estimate of GFR using a formula that includes body weight must be assessed at baseline, during and for at least the first year after treatment.
Monitor serum creatinine and creatinine clearance (see Section 4.2 Dose and Method of Administration, Treatment monitoring (of Lutathera)). Based on the creatinine clearance, Lutathera may require withholding, dose reduction, or permanent discontinuation as described in Table 3 Recommended dose modifications for adverse drug reactions.
Patients with renal impairment at baseline may be at increased risk of toxicity due to increased radiation exposure (see Section 4.2 Dose and Method of Administration). For patients with creatinine clearance < 50 mL/min, an increased risk for transient hyperkalaemia due to the amino acid solution should also be taken into consideration (see Section 4.4 Special Warnings and Precautions for Use, Hyperkalaemia).
Hepatotoxicity. Patients with hepatic metastasis or pre-existing advanced hepatic impairment may be at increased risk of hepatotoxicity due to radiation exposure (see Section 4.2 Dose and Method of Administration).
Transaminases, bilirubin, serum albumin and INR should be monitored during treatment with Lutathera (see Section 4.2 Dose and Method of Administration, Treatment monitoring (of Lutathera)). Based on the severity of hepatotoxicity, Lutathera may require withholding, dose reduction, or permanent discontinuation as described in Table 3 Recommended dose modifications for adverse drug reactions.
Hypersensitivity. Cases of hypersensitivity reactions (including isolated angioedema events) have been reported in the post-marketing setting in patients treated with Lutathera (see Section 4.8 Adverse Effects (Undesirable Effects)). In the event of serious hypersensitivity reactions, the ongoing Lutathera infusion should be discontinued immediately. Appropriate medications and equipment to manage such reactions should be available for immediate use.
Nausea and vomiting. To prevent treatment-related nausea and vomiting, an intravenous bolus of an antiemetic medicinal product should be injected at least 30 minutes prior to the start of amino acid solution infusion to reach the full antiemetic efficacy.
Glucocorticoids should be avoided as preventive antiemetic treatment (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Neuroendocrine hormonal crisis. Crises due to excessive release of hormones or bioactive substances may occur typically within 24 hours following treatment with Lutathera. Patients should be monitored for signs and symptoms of tumour-related hormonal release, including hemodynamic alterations.
Somatostatin analogues, fluids, corticosteroids, and electrolytes should be administered as clinically indicated. In the event of hormonal crisis, overnight hospitalisation of patients should be considered in some cases for observation (e.g. patients with poor pharmacologic control of symptoms).
Radioprotection rules. The healthcare professional should determine when the patient can leave the controlled area of the hospital, i.e. when the radiation exposure to third parties does not exceed regulatory thresholds (see Section 4.4 Special Warnings and Precautions for Use).
Patients should be encouraged to remain hydrated and to urinate frequently before, on the day of and the day after administration of Lutathera to facilitate elimination (e.g. at least 1 glass of water every hour). They should also be encouraged to defecate every day and to use laxative if needed. Urine and faeces should be disposed of according to the national regulations.
Provided the patient's skin is not contaminated, such as from the leakage of the infusion system or because of urinary incontinence, radioactivity contamination is not expected on the skin and in the vomited mass. However, it is recommended that when conducting standard care or examinations with medical devices or other instruments which come into contact with the skin (e.g. electrocardiogram [ECG]), basic protection measures should be observed such as wearing gloves, installing the material/electrode before the start of radiopharmaceutical infusion, changing the material/electrode after measurement, and eventually monitoring the radioactivity of equipment after use.
Before being discharged, the patient should be instructed in the necessary radioprotection rules for interacting with other members of the same household and the general public, and the general precautions the patient must follow during daily activities after treatment (as given in next paragraph and the Consumer Medicine Information) to minimise radiation exposure to others.
The following general recommendations can be considered along with national, local and institutional procedures and regulations following each administration of Lutathera:
Close contact (less than 1 metre) with other people should be limited for 7 days.
For children and/or pregnant women, close contact (less than 1 metre) should be limited to less than 15 minutes per day for 7 days.
Patients should sleep in a separate bedroom from other people for 7 days.
Patients should sleep in a separate bedroom from children and/or pregnant women for 15 days.
Recommended measures in case of extravasation. Disposable waterproof gloves should be worn. The infusion of Lutathera must be immediately ceased and the administration device (catheter, etc.) removed. The nuclear medicine physician and the radiopharmacist should be informed.
All the administration device materials should be kept in order to measure the residual radioactivity and the activity actually administered and the absorbed dose should be determined. The extravasation area should be delimited with an indelible pen and a picture should be taken if possible. It is also recommended to record the time of extravasation and the estimated volume extravasated.
To continue Lutathera infusion, it is mandatory to use a new catheter possibly placing it in a contralateral venous access.
No additional medicinal product can be administered to the same side where the extravasation occurred.
In order to accelerate Lutathera dispersion and to prevent its stagnation in tissue, it is recommended to increase blood flow by elevating the affected arm. Depending on the case, aspiration of extravasation fluid, flush injection of sodium chloride 9 mg/mL (0.9%) solution for injection, or application of warm compresses or a heating pad to the infusion site to accelerate vasodilation should be considered.
Symptoms, especially inflammation and/or pain, should be treated. Depending on the situation, the nuclear medicine physician should inform the patient about the risks linked to extravasation injury and give advice about potential treatment and necessary follow-up requirements. The extravasation area must be monitored until the patient is discharged from the hospital. Depending upon its severity, this event should be declared as an adverse drug reaction.
Patients with urinary incontinence. During the first 2 days following administration of Lutathera, special precautions should be taken with patients with urinary incontinence to avoid spread of radioactive contamination. This includes the handling of any materials possibly contaminated with urine.
Patients with brain metastases. There are no efficacy data in patients with known brain metastases, therefore individual benefit-risk must be assessed in these patients.
Other patients with risk factors. Patients presenting with any of the conditions below are more prone to develop adverse reactions. Therefore, it is recommended to monitor such patients more frequently during the treatment. Please see Table 3 in case of dose modifying toxicity.
Bone metastasis;
Previous oncological radiometabolic therapies with 131I compounds or any other therapy using unshielded radioactive sources;
History of other malignant tumours unless the patient is considered to have been in remission for at least 5 years.
Warnings and precautions regarding the co-administered renal protective amino acid solution. Hyperkalaemia. A transient increase in serum potassium levels may occur in patients receiving amino acid solutions, usually returning to normal levels within 24 hours from the start of the amino acid solution infusion. Patients with reduced creatinine clearance may be at increased risk for transient hyperkalaemia (see Section 4.4 Special Warnings and Precautions for Use, Renal toxicity).
Serum potassium levels must be tested before each administration of amino acid solution. In case of hyperkalaemia, the patient's history of hyperkalaemia and concomitant medication should be checked. Hyperkalaemia must be corrected accordingly before starting the infusion.
In case of pre-existing clinically significant hyperkalaemia, a second monitoring prior to amino acid solution infusion must confirm that hyperkalaemia has been successfully corrected. The patient should be monitored closely for signs and symptoms of hyperkalaemia, e.g. dyspnoea, weakness, numbness, chest pain and cardiac manifestations (conduction abnormalities and cardiac arrhythmias). An electrocardiogram (ECG) should be performed prior to discharging the patient.
Vital signs should be monitored during the infusion regardless of baseline serum potassium levels. Patients should be encouraged to remain hydrated and to urinate frequently before, on the day of and the day after administration (e.g. 1 glass of water every hour) to facilitate elimination of excess serum potassium.
In case hyperkalaemia symptoms develop during amino acid solution infusion, appropriate corrective measures must be taken. In case of severe symptomatic hyperkalaemia, discontinuation of amino acid solution infusion should be considered, taking into consideration the benefit-risk of renal protection versus acute hyperkalaemia.
Heart failure. Due to potential for clinical complications related to volume overload, care should be taken with use of amino acid solutions in patients with severe heart failure defined as class III or class IV in the NYHA (New York Heart Association) classification. Patients with severe heart failure defined as class III or class IV in the NYHA classification should only be treated after a careful benefit-risk assessment, taking into consideration the volume and osmolality of the amino acid solution.
Metabolic acidosis. Metabolic acidosis has been observed with administration of amino acid solutions. Shifts in acid-base balance alter the balance of extracellular-intracellular potassium and the development of acidosis may be associated with rapid increases in plasma potassium.
Effects on laboratory tests. See Section 4.2 Dose and Method of Administration, Treatment monitoring (of Lutathera).
4.5 Interactions with Other Medicines and Other Forms of Interactions
Somatostatin analogues. Somatostatin and its analogues competitively bind to somatostatin receptors and may interfere with the efficacy of Lutathera. Therefore, administration of long-acting somatostatin analogues should be discontinued at least 4 weeks prior to the administration of Lutathera. If necessary, patients may be treated with short-acting somatostatin analogues up to 24 hours preceding Lutathera administration.
Glucocorticoids. There is some evidence that glucocorticoids can induce down-regulation of subtype 2 somatostatin receptors (SST2). Repeated administration of high doses of glucocorticoids should be avoided during treatment with Lutathera. Patients with history of chronic use of glucocorticoids should be carefully evaluated for sufficient somatostatin receptor expression. It is not known whether the intermittent use of glucocorticoids for the prevention of nausea and vomiting during Lutathera administration could induce SST2 down-regulation. As a matter of caution, glucocorticoids should also be avoided as preventive antiemetic treatment. In the event that the treatment administered for the prevention of nausea and vomiting before the amino acid solution infusion proves insufficient, a single glucocorticoid dose can be used, provided it is not given before initiating or within one hour after the end of Lutathera infusion.
In vitro evaluation of drug interaction potential. Metabolic and transporter based interaction. In vitro drug interaction studies performed with non-radioactive lutetium (175Lu) oxodotreotide showed an absence of significant inhibitory or induction effects on human CYP450 enzymes (CYP1A2, 2B6, 2C9, 2C19 or 2D6), no potential interactions with P-glycoprotein (efflux transporter), as well as OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, and BCRP transporters. Therefore, Lutathera has a low probability of causing clinically relevant metabolism- or transporter-mediated interactions.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. No animal studies were conducted to determine the effects of lutetium (177Lu) oxodotreotide on male and female fertility. Lutathera may cause infertility in males and females. The recommended cumulative dose of 29.6 GBq of Lutathera results in a radiation absorbed dose to the testes and ovaries within the range where temporary or permanent infertility may occur following external beam radiotherapy.
Genetic consultation is recommended if the patient wishes to have children after treatment. Cryopreservation of sperm or eggs can be discussed as an option for patients before treatment.
Pregnancy testing. The pregnancy status of females of reproductive potential must be verified prior to initiating treatment with Lutathera (see Section 4.2 Dose and Method of Administration).
Contraception. Females. Lutathera can cause fetal harm when administered to a pregnant woman. Female patients of reproductive potential should be advised to use effective contraception during treatment and for 7 months after the last dose of Lutathera.
Males. Based on its mechanism of action, male patients with female partners of reproductive potential should be advised to use effective contraception during treatment and for 4 months after the last dose of Lutathera.
Use in pregnancy. (Category X)
Lutathera is contraindicated in patients with established or suspected pregnancy or when pregnancy has not been excluded (see Section 4.3 Contraindications). Based on its mechanism of action, Lutathera can cause fetal harm when administered to a pregnant woman.
There are no available data on Lutathera use in pregnant women. No animal studies using lutetium (177Lu) oxodotreotide have been conducted to evaluate its effect on female reproduction and embryo-fetal development; however, Lutathera being a radiopharmaceutical has the potential to cause fetal harm. Pregnant women should be advised of the potential risk to a fetus.
Use in lactation. There are no data on the presence of lutetium (177Lu) oxodotreotide in human milk, or its effects on the breast-fed child or milk production. No lactation studies in animals were conducted. Because of the potential risk for serious adverse drug reactions in breast-fed children, women receiving Lutathera should be advised to not breast-feed. If Lutathera treatment is started during breast-feeding, breast-feeding should be discontinued.
4.7 Effects on Ability to Drive and Use Machines
The effects of this medicine on a person's ability to drive and use machines were not assessed as part of its registration.
4.8 Adverse Effects (Undesirable Effects)
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
Summary of the safety profile. The overall safety profile of Lutathera is based on data from patients from clinical studies (NETTER-1 phase III and ERASMUS phase I/II) and from compassionate use programs.
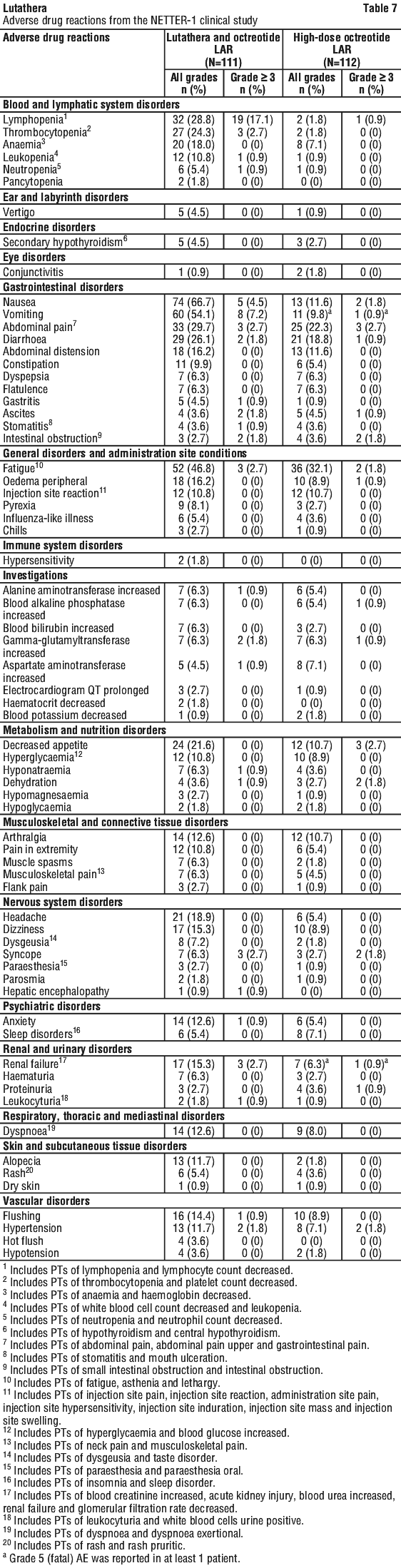
The safety data and frequency of the adverse drug reactions reported below are based on NETTER-1 (n=111) and ERASMUS (n=811).
Very common ADRs (at frequency ≥ 20%) were: nausea (64.3%), vomiting (52.7%), fatigue (48.2%), abdominal pain (32.1%), lymphopenia (28.6%), diarrhoea (25.9%), thrombocytopenia (25.9%), decreased appetite (20.5%).
Nausea and vomiting occurred mainly at the beginning of the infusion. The causality of nausea/vomiting is confounded by the emetic effect of the concomitant amino acid solution administered for renal protection.
At the time of the NETTER-1 final analysis, after a median follow-up duration of 76 months in each study arm, the safety profile remained consistent with that previously reported.
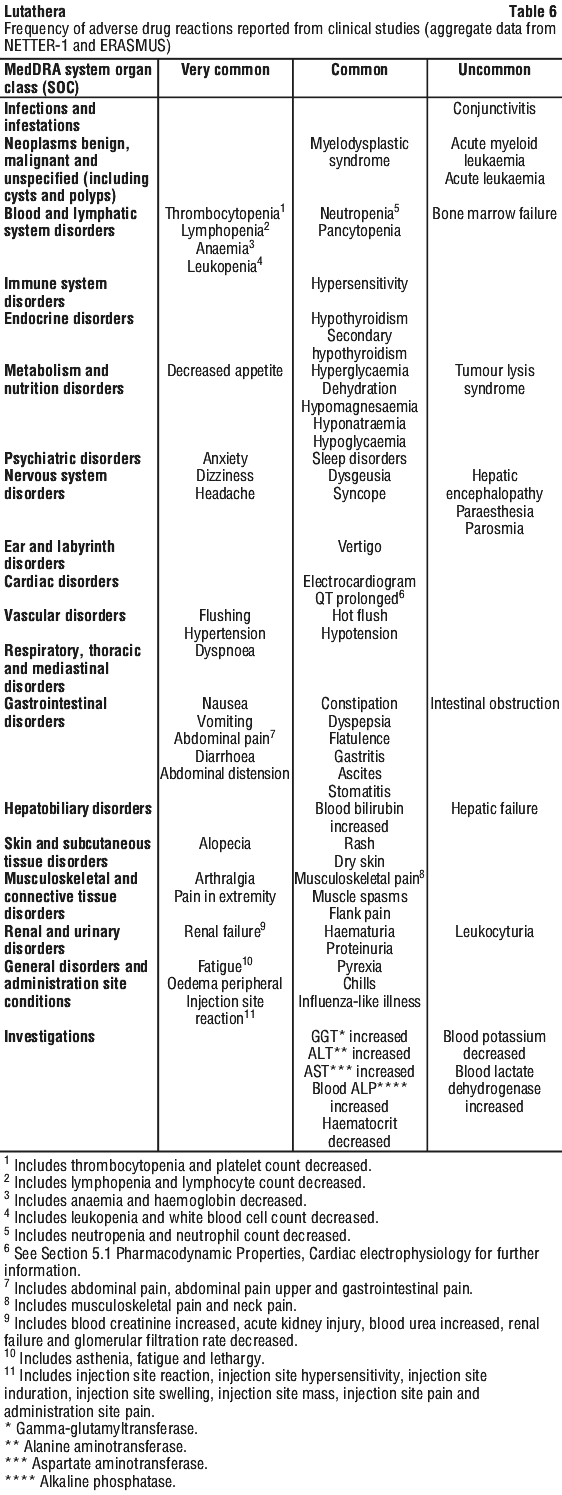
Tabulated summary of adverse drug reactions from clinical studies. Adverse drug reactions from clinical studies (Table 6) are listed by MedDRA system organ class. Within each system organ class, the adverse drug reactions are ranked by frequency, with the most frequent reactions first. In addition, the corresponding frequency category for each adverse drug reaction is based on the following convention (CIOMS III): very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000). See Table 7.

In NETTER-1, platelet nadir occurred at a median of 5.1 months following the first dose. Of the 59 patients who developed thrombocytopenia, 68% had platelet recovery to baseline or normal levels. The median time to platelet recovery was 2 months. Fifteen of the nineteen patients in whom platelet recovery was not documented had post-nadir platelet counts.
Among these 15 patients, 5 improved to Grade 1, 9 to Grade 2, and 1 to Grade 3.
Renal toxicity. Lutetium (177Lu) oxodotreotide is excreted by the kidney. The long-term trend of progressive glomerular filtration function deterioration demonstrated in the clinical studies confirms that Lutathera-related nephropathy is a chronic kidney disease that develops progressively over months or years after exposure. An individual benefit-risk assessment is recommended prior to treatment with Lutathera in patients with mild or moderate renal impairment.
The use of Lutathera is contraindicated in patients with kidney failure with creatinine clearance < 30 mL/min.
4.9 Overdose
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
Overdose is unlikely with Lutathera as this medicinal product is supplied as a "single-dose" and "ready-to-use" product containing a predefined amount of radioactivity and should be used by or under the control of physicians who are qualified by specific training and experience in the safe use and handling of radiopharmaceuticals. In the event of overdose, an increase in the frequency of the adverse drug reactions related to radiotoxicity is expected.
In the event of administration of a radiation overdose with Lutathera, the radiation absorbed dose to the patient should be reduced where possible by increasing the elimination of the radionuclide from the body by frequent micturition or by forced diuresis and frequent bladder voiding during the first 48 hours after infusion. It might be helpful to estimate the effective radiation dose that was applied.
Haematological monitoring, including white blood cell counts (with differential counts), platelets, and haemoglobin, and blood chemistry monitoring, including serum creatinine and blood glucose should be performed every week for the next 10 weeks.
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
At the concentration used (about 10 micrograms/mL in total, for both free and radiolabelled forms), the peptide oxodotreotide does not exert any clinically relevant pharmacodynamic effect.
Pharmacotherapeutic group, ATC. Pharmacotherapeutic group: Therapeutic radiopharmaceuticals, other therapeutic radiopharmaceuticals, ATC code: V10XX04.
Mechanism of action. Lutetium (177Lu) oxodotreotide has a high affinity for subtype 2 somatostatin receptors (SST2). It binds to malignant cells which express SST2.
Lutetium-177 is a beta-minus emitting radionuclide with a maximum penetration range in tissue of 2.2 mm (mean penetration range of 0.67 mm), causing death of the targeted tumour cells with a limited effect on neighbouring normal cells.
Cardiac electrophysiology. The ability of Lutathera to prolong the QTc interval at the recommended dose was assessed in an open-label study in 18 patients with somatostatin receptor-positive midgut carcinoid tumours. Single doses of Lutathera resulted in mean (90% CI) QTcF change from baseline of 2.8 (-1.5, 7.2) msec during the first 2 hours, 4.2 (-2.5, 11.0) msec at 4 hours, 10 (4.0, 15.9) msec at 8 hours and 11.1 (5.3, 16.8) msec at 24 hours. No subject had a QTcF value exceeding 480 msec or ΔQTcF > 60 msec. A concentration dependent increase in QTc was not detected. No clinically relevant changes in the mean QTc interval (i.e. > 20 msec) were detected.
Clinical trials. NETTER-1 study. The NETTER-1 phase III study was a multicentre, stratified, open-label, randomised, comparator-controlled, parallel-group study comparing treatment with Lutathera (4 doses of 7400 MBq, one dose every 8 weeks [± 1 week]) co-administered with an amino acid solution and best supportive care (octreotide long-acting release [LAR] 30 mg after each Lutathera dose and every 4 weeks after completion of Lutathera treatment for symptom control, replaced by short-acting octreotide in the 4-week interval before Lutathera administration) to high-dose octreotide LAR (60 mg every 4 weeks) in patients with inoperable, progressive, somatostatin receptor-positive, midgut carcinoid tumours. The primary endpoint for the study was progression-free survival (PFS) evaluated by response evaluation criteria in solid tumours (RECIST v1.1), based on blinded independent review committee (IRC) assessment. Secondary efficacy endpoints included objective response rate (ORR), overall survival (OS), time to tumour progression (TTP), safety and tolerability of the medicinal product, and health-related quality of life (HRQoL).
At the time of the primary analysis, 229 patients were randomised (1:1). Randomisation was stratified by OctreoScan tumour uptake score (Grade 2, 3 or 4) and the length of time that patients had been on the most recent constant dose of octreotide prior to randomisation (≤ 6 or > 6 months).
Demographic and baseline disease characteristics were balanced between the treatment arms. Of the 208 patients, whose race/ethnicity was reported, 90% were White, 5% were Black, and 4% were Hispanic or Latino. The median age was 64 years (28 to 87 years); 51% were male, and 96% had metastatic disease in the liver. The median Karnofsky performance score was 90 (60 to 100), 74% received a constant dose of octreotide for > 6 months and 12% received prior treatment with everolimus. Sixty-nine percent (69%) of patients had Ki67 expression in ≤ 2% of tumour cells, 77% had CgA > 2 times the upper limit of normal (ULN), 65% had 5-HIAA > 2 times ULN, and 65% had alkaline phosphatase ≤ ULN.
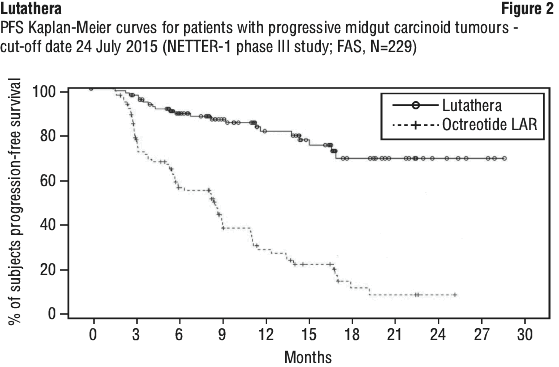
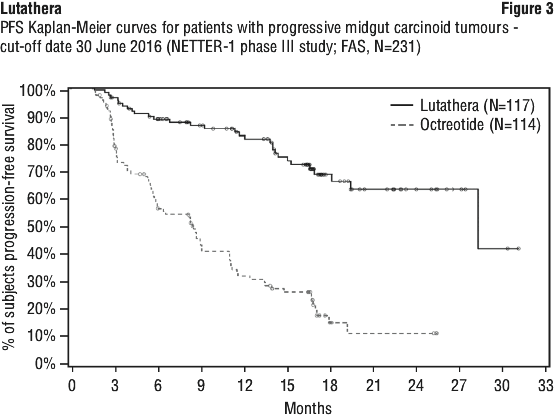
At the time of the primary PFS analysis (cut-off date 24 July 2015), the number of centrally confirmed disease progressions or deaths was 21 events in the Lutathera arm and 70 events in the high-dose octreotide LAR arm (Table 9). PFS differed significantly (p < 0.0001) between the treatment arms. The median PFS for the Lutathera arm was not reached at the cut-off date, whereas the median PFS for the high-dose octreotide LAR arm was 8.5 months. The hazard ratio for the Lutathera arm compared to the high-dose octreotide LAR arm was 0.18 (95% CI: 0.11; 0.29), indicating 82% reduction in the risk of disease progression or death in favour of the Lutathera arm.


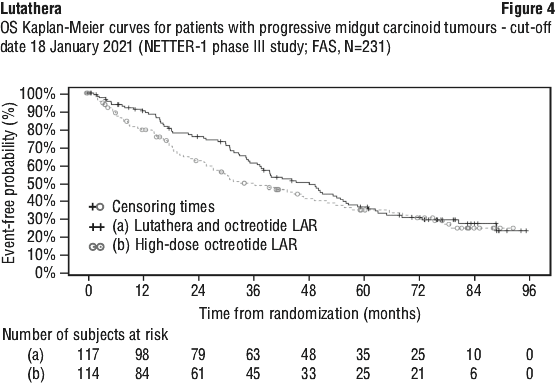
At the time of the final OS analysis, which occurred 5 years after the last patient was randomised (N=231, cut-off date 18 January 2021), the median follow-up duration was 76 months in each study arm. There were 73 deaths in the Lutathera arm (62.4%) and 69 deaths in the high-dose octreotide LAR arm (60.5%), yielding a HR of 0.84 (95% CI: 0.60; 1.17; unstratified Log-rank test p=0.3039, two-sided) in favour of the Lutathera arm. The median OS was prolonged by a clinically relevant extent of 11.7 months in patients randomised to the Lutathera arm compared to patients randomised to high-dose octreotide LAR, with a median OS of 48.0 months (95% CI: 37.4; 55.2) and 36.3 months (95% CI: 25.9; 51.7), respectively. The final OS results did not reach statistical significance [15,16,18]. In the high-dose octreotide LAR arm, 22.8% of patients received subsequent radioligand therapy (including lutetium (177Lu) oxodotreotide) within 24 months of randomisation, and 36% of patients received subsequent radioligand therapy by the final OS cut-off date, which along with other factors may have influenced the OS in this subset of patients.
The OS Kaplan-Meier graph for the FAS at the cut-off date 18 January 2021 is depicted in Figure 4.
ERASMUS study. The efficacy of Lutathera in patients with foregut (including bronchial), midgut, and hindgut neuroendocrine tumours (NETs) was assessed in 360 patients in the ERASMUS study. In ERASMUS, Lutathera was initially provided under a general peptide receptor radionuclide therapy protocol at a single site in the Netherlands. A subsequent Lutathera specific protocol written eight years after study initiation did not describe a specific sample size or hypothesis testing plan but allowed for retrospective data collection. A total of 1,214 patients received Lutathera in ERASMUS, of whom 578 patients had baseline tumour assessment. Of the 578 patients with baseline tumour assessment, 360 (62%) patients had gastroenteropancreatic (GEP) and bronchial NETs and long-term follow-up. Out of the 360 patients (full analysis set, FAS), 183 had midgut tumours, 133 had pancreatic tumours, 19 patients had bronchial tumours, 13 had hindgut tumours and 12 had foregut tumours (other than bronchial and pancreatic). The median age in the FAS was 60 years (30 to 85 years), 51% were male, 71% had a baseline Karnofsky performance status ≥ 90, 51% had progressed within 12 months of treatment, and 7% had received prior chemotherapy. Fifty-two percent (52%) of patients received a concomitant long-acting release somatostatin analogue.
Lutathera 7400 MBq (200 mCi) was administered every 6 to 13 weeks for up to 4 doses concurrently with the recommended amino acid solution. The median dose of Lutathera was 29600 MBq (800 mCi). The major efficacy outcome was investigator-assessed ORR as an aggregate of the best overall response (BOR) in the 5 subtypes of NETs. Patients in the FAS had their tumours assessed using either the RECIST v1.1 criteria (145 patients, 40%) or the SWOG assessment which was retrospectively algorithmically converted to RECIST v1.1 (215 patients, 60%).
The overall investigator-assessed ORR was 45% (95% CI: 40; 50) and the median duration of response (DoR) was 22.9 months (95% CI: 17; 25) (Table 11). The observed ORR was highest for pancreatic NET patients (61%, 95% CI: 52; 69) and lowest for midgut NET patients (33%, 95% CI: 27; 41). In the subset of 145 patients who were evaluated by the investigators using RECIST v1.1 criteria, the ORR was 41% (95% CI: 33; 50), and median DoR was 35 months (95% CI: 17; 38), and in the subset of 215 patients who were evaluated by the investigators using the converted SWOG criteria, the ORR was 47% (95% CI: 41; 54), and median DoR was 18.5 months (95% CI: 15; 24).

5.2 Pharmacokinetic Properties
The pharmacokinetics of lutetium (177Lu) oxodotreotide have been characterised in patients with progressive, somatostatin receptor-positive neuroendocrine tumours.
Absorption. The mean blood exposure (AUC) of lutetium (177Lu) oxodotreotide at the recommended dose is 41 nanogram.h/mL [coefficient of variation (CV) 36%]. The mean maximum blood concentration (Cmax) for lutetium (177Lu) oxodotreotide is 10 nanogram/mL (CV 50%), which generally occurred at the end of the Lutathera infusion.
Distribution. The mean volume of distribution (Vz) for lutetium (177Lu) oxodotreotide is 460 L (CV 54%). The non-radioactive form (lutetium (175Lu) oxodotreotide) is 43% bound to human plasma proteins.
Within 4 hours after administration, lutetium (177Lu) oxodotreotide distributes in kidneys, tumour lesions, liver, spleen, and, in some patients, pituitary gland and thyroid. In a limited dataset (n=4), the co-administration of amino acids reduced the median radiation dose to the kidneys by 47% (34% to 59%) and increased the mean beta-phase blood clearance of lutetium (177Lu) oxodotreotide by 36%.
Metabolism. Lutetium (177Lu) oxodotreotide does not undergo hepatic metabolism.
Based on the analysis of urine samples of 20 patients included in the NETTER-1 phase III Dosimetry, pharmacokinetic and ECG sub-study, lutetium (177Lu) oxodotreotide is poorly metabolised and is excreted mainly as intact compound by renal route.
Excretion. The mean clearance (CL) is 4.5 L/h (CV 31%) for lutetium (177Lu) oxodotreotide.
The mean terminal blood elimination half-life is 71 (± 28) hours for lutetium (177Lu) oxodotreotide.
Lutetium (177Lu) oxodotreotide is primarily eliminated renally with cumulative excretion of 44% within 5 hours, 58% within 24 hours, and 65% within 48 hours following Lutathera administration. Prolonged elimination of lutetium (177Lu) oxodotreotide in the urine is expected; however, based on the half-life of lutetium-177 and terminal blood elimination half-life of lutetium (177Lu) oxodotreotide, greater than 99% of the administered radioactivity will be eliminated within 14 days after administration of Lutathera (see Section 4.4 Special Warnings and Precautions for Use).
Special populations. Geriatric patients (75 years of age or above). The pharmacokinetics profile in geriatric patients (≥ 75 years) has not been established. No data are available.
5.3 Preclinical Safety Data
Genotoxicity. Genotoxicity studies showed that non-radioactive lutetium (175Lu) oxodotreotide formulation does not induce mutation at the TK locus of L5178Y mouse lymphoma cells in vitro, nor reverse mutation in Salmonella typhimurium or Escherichia coli in the absence or presence of S9 metabolic activation. However, lutetium (177Lu) oxodotreotide is radioactive, and radiation is a mutagen.
Carcinogenicity. Mutagenicity studies and long-term carcinogenicity studies have not been carried out with lutetium (177Lu) oxodotreotide; however, radiation is a carcinogen and mutagen.
6 Pharmaceutical Particulars
6.1 List of Excipients
Acetic acid (0.48 mg/mL), sodium acetate (0.66 mg/mL), gentisic acid (0.63 mg/mL), ascorbic acid (2.80 mg/mL), pentetic acid (0.05 mg/mL), sodium chloride (6.85 mg/mL), sodium hydroxide (0.64 mg/mL), water for injection(s) (add to 1 mL).
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those mentioned in Section 4.2 Dose and Method of Administration.
6.3 Shelf Life
72 hours from the date and time of calibration.
6.4 Special Precautions for Storage
Store below 25°C. Do not freeze Lutathera.
Store in the original package to protect from ionising radiation (lead shielding).
Storage of radiopharmaceuticals should be in accordance with national regulations on radioactive materials.
Lutathera must be kept out of the reach and sight of children.
6.5 Nature and Contents of Container
Clear, colourless type I glass vial, closed with a bromobutyl rubber stopper and aluminium cap.
Each vial contains a volume that ranges from 20.5 to 25.0 mL of solution, corresponding to a radioactivity of 7400 MBq ± 10% at the date and time of infusion.
The vial is enclosed within a lead shielded container and placed in a plastic sealed container.
6.6 Special Precautions for Disposal
Any unused medicinal product or waste material should be disposed of in accordance with national regulations.
Lutetium-177 for Lutathera may be prepared using two different sources of stable nuclides (either lutetium-176 or ytterbium-176) resulting in different waste management. The user must consult the documentation provided before using Lutathera to ensure appropriate waste management.
6.7 Physicochemical Properties
Chemical structure.
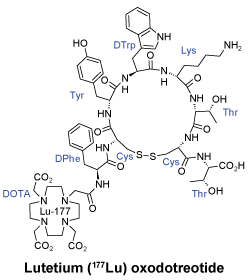
CAS number. 437608-50-9.
7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription only medicine.
Date of First Approval
17 November 2025
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.