Luxturna
Brand Information
| Brand name | Luxturna |
| Active ingredient | Voretigene neparvovec |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Luxturna.
Summary CMI
Luxturna®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
▼ This medicine is new or being used differently. Please report side effects. See the full CMI for further details.
1. Why am I given Luxturna?
Luxturna contains the active substance Voretigene neparvovec. Luxturna is used to treat certain genetic diseases that damage the retina (the layer at the back of the eye that detects light) caused by RPE65 mutations.
For more information, see Section 1. Why am I given Luxturna? in the full CMI.
2. What should I know before Luxturna is given?
Do not use if you have ever had an allergic reaction to Luxturna or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding. For more information, see Section 2. What should I know before Luxturna is given? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with Luxturna and affect how it works. A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How will you be given Luxturna?
- Your doctor will prescribe you certain medicines prior to treatment with Luxturna.
- Follow the instructions given by your doctor carefully.
More instructions can be found in Section 4. How will you be given Luxturna? in the full CMI.
5. What should I know when Luxturna is given?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Drinking alcohol |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know when Luxturna is given? in the full CMI.
6. Are there any side effects?
Common side effects include eye pain, eye irritation, vision problems, increased sensitivity to light, feeling that something is in your eye(s), redness of the eye(s), eye swelling, headache, blurred vision.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
▼ This medicine is subject to additional monitoring. This will allow quick identification of new safety information. You can help by reporting any side effects you may get. You can report side effects to your doctor, or directly at www.tga.gov.au/reporting-problems.
Luxturna® Concentrate and solvent for solution for injection
Active ingredient: VORETIGENE NEPARVOVEC
Consumer Medicine Information (CMI)
This leaflet provides important information about using Luxturna. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using Luxturna.
Where to find information in this leaflet:
1. Why am I given Luxturna?
2. What should I know before Luxturna is given?
3. What if I am taking other medicines?
4. How will you be given Luxturna?
5. What should I know when Luxturna is given?
6. Are there any side effects?
7. Product details
1. Why am I given Luxturna?
Luxturna contains the active substance Voretigene neparvovec. It is a modified virus containing a copy of the RPE65 gene.
Luxturna is used to treat certain genetic diseases that damage the retina (the layer at the back of the eye that detects light) caused by RPE65 mutations. These mutations prevent the body's ability to produce a protein required for vision, that results in vision loss and eventually blindness.
Your doctor will assess if you have enough retinal cells for Luxturna to work properly.
Voretigene neparvovec is a modified virus that carries a copy of the RPE65 gene. Once injected into the eye, it delivers this gene to retinal cells. This enables the retina to produce essential proteins needed for vision. The virus that is used to deliver the gene does not cause disease in humans.
2. What should I know before Luxturna is given?
Warnings
Do not use Luxturna:
- If you are allergic to Voretigene neparvovec, or any of the ingredients listed at the end of this leaflet. Always check the ingredients to make sure you can use this medicine. Symptoms of an allergic reaction may include shortness of breath, difficulty breathing, swelling of the face, lips, tongue or other parts of the body, rash, hives.
- If you suffer from any eye infections or eye swelling. Symptoms include redness of the eye, eye swelling, eye pain, sensitivity to light.
Check with your doctor if you:
- have any other medical conditions.
- take any medicines for any other condition.
- suffer from increased eye pressure.
- have diseases of cornea with symptoms such as eye pain, feeling that something is in your eye(s), sensitivity to light.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
Avoid the use of Luxturna during pregnancy.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
Your doctor will advise you regarding the possible risks and benefits of using Luxturna during breastfeeding.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect Luxturna.
4. How will you be given Luxturna?
How much to use
Before you receive Luxturna:
- Your doctor will prescribe you certain medicines prior to treatment with Luxturna.
- Follow the instructions given by your doctor carefully.
How you will be given Luxturna:
- You will be given Luxturna in an operating room by a surgeon experienced in performing eye surgery. It will be injected into your eye.
- Luxturna is given under anaesthesia. Your doctor will talk to you about the process of anaesthesia.
- Your doctor will carry out eye surgery, and then inject Luxturna directly into the retina, the thin light-sensing layer at the back of the eye.
- This surgery will be repeated on your other eye at least 6 days afterwards.
- You will need to stay for post-operative observation for a few hours after each procedure to monitor your recovery and watch for any side effects from either the surgery or anaesthesia.
If you use too much Luxturna
It is unlikely that you will be given too much Luxturna as it will be given to you by a doctor. If this occurs, your doctor will treat the symptoms as needed.
5. What should I know when Luxturna is given?
Things you should do
- Avoid air travel or travel to high elevations until your doctor tells you to.
- Some of the medicine may be present in your tears. You and your caregiver should wear gloves during dressing changes, especially if you are pregnant, breastfeeding or have any problems with your immune system.
- You and your caregiver should continue to wear gloves when disposing of the dressings and other waste material.
Call your doctor straight away if you:
- develop any signs of an allergic reaction.
- develop any symptoms of an eye infection or swelling, symptoms such as redness of the eye, eye swelling, eye pain, sensitivity to light.
- you see flashes or floaters, or if you notice any worsening or blurred vision.
- have any signs or symptoms such as flashes of light in your vision, reduced vision or blurred or distorted vision e.g. seeing straight lines as wavy.
Remind any doctor, dentist, pharmacist or specialist you visit that you have used Luxturna.
Things you should not do
- Do not donate blood, organs, tissues and cells for transplantation after treatment with Luxturna. This is because Luxturna is a gene therapy product.
- Avoid swimming because of an increased risk of infection in the eye. You may resume swimming after a minimum of one to two weeks, on the advice of your doctor.
- Do not use Luxturna in children under 4 years of age.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how Luxturna affects you and follow the advice given by your doctor.
Luxturna may cause temporary vision problems.
Do not drive or use heavy machines until your vision has recovered sufficiently.
Drinking alcohol
Tell your doctor if you drink alcohol.
Looking after your medicine
- Luxturna will be stored by a healthcare professional at your healthcare facility.
- The concentrate and solvent must be stored and transported frozen at less than or equal to minus 65°C. Once thawed, the medicine should not be re-frozen and should be left at room temperature (below 25°C).
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
Eye Problems:
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
Eye problems:
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What Luxturna contains
| Active ingredient (main ingredient) | Voretigene neparvovec Luxturna concentrate for subretinal injection contains 5 x 1012 vector genomes (vg) per mL. The concentrate (0.5 mL extractable volume in a single-dose 2 mL vial) requires a 1:10 dilution prior to administration. After dilution, each dose contains 1.5 x 1011 vg in a deliverable volume of 0.3 mL. |
| Other ingredients (inactive ingredients) | Concentrate: Sodium chloride, monobasic sodium phosphate (for pH adjustment), dibasic sodium phosphate (for pH adjustment), poloxalene, water for injections. Diluent: Sodium chloride, monobasic sodium phosphate (for pH adjustment), dibasic sodium phosphate (for pH adjustment), poloxalene, water for injections. |
| Potential allergens | NA |
Do not take this medicine if you are allergic to any of these ingredients.
What Luxturna looks like
Luxturna is a clear, colourless preservative-free liquid.
It is supplied in 0.5 mL extractable volume of concentrate in 2 mL cyclic olefin polymer vial with a chlorobutyl rubber stopper sealed in place with an aluminium flip-off seal.
1.7 mL extractable volume of solvent in a 2 mL cyclic olefin polymer vial with a chlorobutyl rubber stopper sealed in place with an aluminium flip-off seal.
Each foil pouch includes a carton containing 1 vial of concentrate and 2 vials of solvent.
Australian registration number: AUST R: 318929.
Who distributes Luxturna
This product is supplied in Australia by:
Novartis Pharmaceuticals Australia Pty Limited
54 Waterloo Road
Macquarie Park NSW 2113
Telephone no. 1800 671 203.
Website:
www.novartis.com.au
® = Registered Trademark.
© Copyright 2025.
© Novartis Pharmaceuticals Australia Pty Limited 2025
This leaflet was prepared in December 2025.
Internal code
lux111125c_V2 based on PI lux111125i
Brand Information
| Brand name | Luxturna |
| Active ingredient | Voretigene neparvovec |
| Schedule | S4 |
▼ This medicinal product is subject to additional monitoring in Australia. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse events at www.tga.gov.au/reporting-problems.
MIMS Revision Date: 01 January 2026
1 Name of Medicine
Voretigene neparvovec (recombinant adeno-associated virus 2 vector AAV2-Hrpe65V2).
2 Qualitative and Quantitative Composition
Voretigene neparvovec is a gene transfer vector that employs an adeno-associated viral vector serotype 2 (AAV2) capsid as a delivery vehicle for the human retinal pigment epithelium 65 kDA protein (hRPE65) cDNA to the retina. Voretigene neparvovec is derived from naturally occurring AAV using recombinant DNA techniques.
Luxturna concentrate for subretinal injection contains 5 x 1012 vector genomes (vg) per mL.
Luxturna is supplied in a 2 mL single-dose vial containing 0.5 mL extractable volume that requires a 1:10 dilution prior to administration.
After dilution, each dose contains 1.5 x 1011 vg in a deliverable volume of 0.3 mL.
For the full list of the excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Concentrated solution for subretinal injection.
Both the concentrate and the diluent are clear, colourless preservative-free liquids.
4 Clinical Particulars
4.1 Therapeutic Indications
Luxturna is indicated for the treatment of patients with inherited retinal dystrophy caused by pathological biallelic RPE65 mutations and who have sufficient viable retinal cells as determined by the treating physician.
Pathological mutations of RPE65 should be confirmed by a National Association of Testing Authorities (NATA) or International Laboratory Accreditation Cooperation (ILAC) accredited laboratory.
4.2 Dose and Method of Administration
Dosage. Treatment should be initiated and administered by a retinal surgeon experienced in performing macular surgery.
Patients will receive a single dose of 1.5 x 1011 vg of Luxturna in each eye. Each dose will be delivered into the subretinal space in a total volume of 0.3 mL. The individual administration procedure to each eye is performed on separate days within a close interval, but no fewer than 6 days apart.
Immunomodulatory regimen. Prior to initiation of the immunomodulatory regimen and prior to administration of Luxturna, the patient must be checked for symptoms of active infectious disease of any nature, and in case of such infection the start of treatment must be postponed until after the patient has recovered.
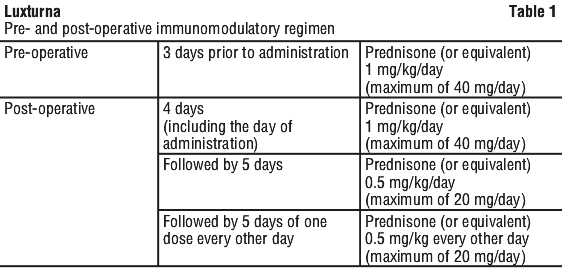
Starting 3 days prior to the administration of Luxturna to the first eye, it is recommended that an immunomodulatory regimen is initiated following the schedule outlined in Table 1. Initiation of the immunomodulatory regimen for the second eye should follow the same schedule and supersede completion of the immunomodulatory regimen of the first eye.
Surgeons should advise patients on the appropriate means of disposing of dressings, waste materials with tears and nasal secretions in the 14 days post-surgery. Patients and caregivers should be advised to wear gloves during dressing changes and place waste materials in a sealed bag for disposal. Patients should be advised to dispose of the sealed bags either through clinical biohazard containers but, if not available, the sealed bags may be placed in normal household waste.
Precautions to be taken before manipulation or administering the medicinal product. This medicinal product contains genetically modified organisms. Personal equipment (to include laboratory coat, safety glasses and gloves) should be worn while preparing or administering voretigene neparvovec (see Section 6.6 Special Precautions for Disposal).
Administration. Prepare Luxturna within 4 hours of administration using sterile technique under aseptic conditions in a Class II vertical laminar flow biological safety cabinet (BSC). Below is the list of items required for dilution of the concentrate and preparation of the administration syringe:
One single-dose vial of Luxturna;
Two vials of Diluent;
One 3-mL sterile syringe;
One 20G 1-inch sterile needle;
Three 1-mL sterile syringes;
Three 27G ½-inch sterile needles;
Two sterile syringe caps;
One 10-mL sterile empty glass vial;
One sterile utility drape;
One sterile plastic bag;
Two sterile labels for administration syringes;
One sterile plain label;
Two sterile skin markers.
Dilution of Luxturna. 1. Thaw one single-dose vial of Luxturna and two vials of Diluent at temperatures below 30°C (room temperature).
2. Mix the contents of the thawed Diluent vials by gently inverting them approximately 5 times.
3. Inspect the Diluent vials. If particulates, cloudiness, or discolouration are visible, do not use the vial(s); new vial(s) of Diluent should be used.
4. Obtain a 3-mL sterile syringe, a 20G 1-inch sterile needle, and a 10-mL sterile empty glass vial.
5. Using the 3-mL syringe with 20G 1-inch needle, transfer 2.7 mL of Diluent to the 10-mL glass vial. Dispose of the needle and syringe in an appropriate container.
6. Mix the contents of the thawed Luxturna single-dose vial by gently inverting approximately 5 times.
7. Inspect the Luxturna single-dose vial. If particulates, cloudiness, or discolouration are visible, do not use the vial; a new single-dose vial of Luxturna should be used.
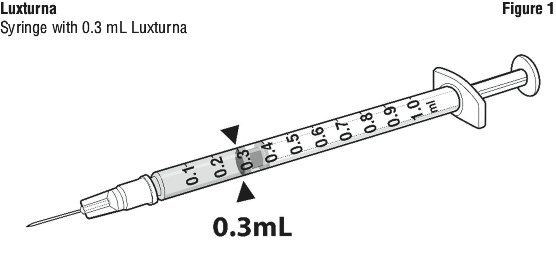
8. Obtain a 1-mL sterile syringe and 27 G ½-inch sterile needle. Draw 0.3 mL of Luxturna into a 1-mL sterile syringe with a 27G ½-inch sterile needle (Figure 1).
10. Using the sterile plain label and sterile skin marker, label the 10-mL glass vial containing the diluted Luxturna as follows: 'Diluted Luxturna'.
11. Remove all items from the BSC except the glass vial labeled 'Diluted Luxturna'.
12. Re-sanitise the BSC prior to the next steps and place the glass vial to the left side in the BSC.
Preparation of Luxturna for injection. To keep the syringes sterile, two operators are required for transfer of the contents of the 10 mL glass vial labeled 'Diluted Luxturna' into each of two sterile 1-mL syringes.
13. Place a sterile utility drape, a sterile plastic bag, and two sterile labels into the BSC.
14. Place the sterile drape near the Primary Operator on the right side of the sanitised BSC surface, away from the diluted Luxturna.
15. The Secondary Operator unwraps two 1-mL syringes, two 27G ½-inch needles, one sterile skin marker and two syringe caps in the BSC, ensuring that the Primary Operator touches only sterile surfaces while transferring the items onto the sterile drape.
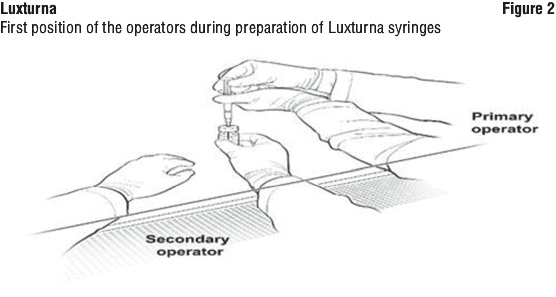
16. The Secondary Operator changes to a new pair of sterile gloves and stands or sits to the left of the Primary Operator. The Secondary Operator holds the 10-mL glass vial containing the diluted Luxturna (Figure 2).
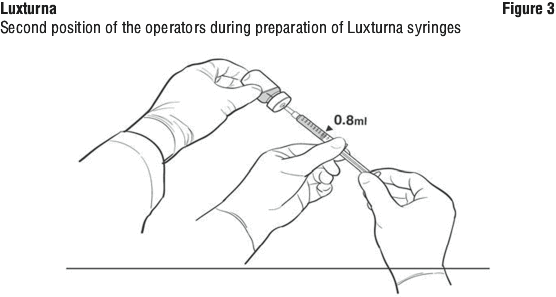
19. The Primary Operator repeats Steps 17 and 18 to prepare a total of two administration syringes. Label the first syringe "Diluted Luxturna" and label the second syringe "Back-up Diluted Luxturna" using the sterile skin marker. The second syringe will serve as a back-up for the surgeon performing the subretinal administration procedure. Discard the back-up syringe after surgery if not used.
20. Inspect both syringes. If particulates, cloudiness, or discolouration are visible, do not use the syringe.
21. Place the syringes into the sterile plastic bag after visual inspection and seal the bag.
22. Place the sterile plastic bag with syringes containing diluted Luxturna into an appropriate secondary container (e.g. hard plastic cooler) for delivery to the surgical suite at room temperature.
Administration. Luxturna should be administered in the surgical suite under controlled aseptic conditions by a surgeon experienced in performing intraocular surgery. In addition to the syringe containing the diluted Luxturna, the following items are required for administration (Figure 4):
Subretinal injection cannula with a polyamide micro tip with an inner diameter of 41 gauge.
Extension tube made of polyvinyl chloride no longer than 15.2 cm (6") in length and with an inner diameter no greater than 1.4 mm.
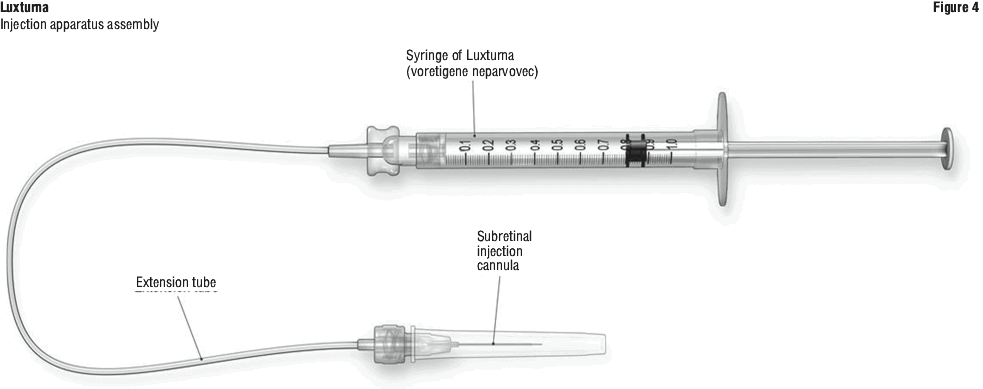
1. After confirming the availability of Luxturna, dilate the eye and give adequate anesthesia to the patient.
2. Administer a topical broad spectrum microbiocide to the conjunctiva, cornea and eyelids prior to surgery.
3. Inspect Luxturna prior to administration. If particulates, cloudiness, or discolouration are visible, do not use the product.
4. Connect the syringe containing the diluted Luxturna to the extension tube and subretinal injection cannula. To avoid excess priming volume, the extension tube should not exceed 15.2 cm in length and 1.4 mm in inner diameter. Inject the product slowly through the extension tube and the subretinal injection cannula to eliminate any air bubbles.
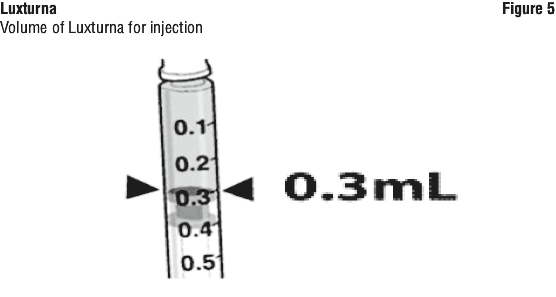
5. Confirm the volume of product available in the syringe for injection, by aligning the plunger tip with the line that marks 0.3 mL (Figure 5).
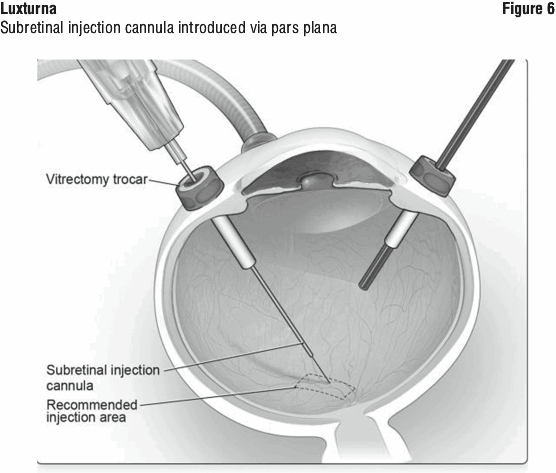
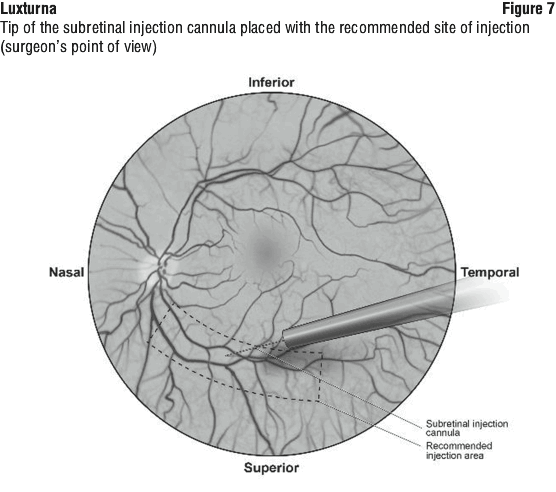
7. Under direct visualisation, place the tip of the subretinal injection cannula in contact with the retinal surface. The recommended site of injection is located along the superior vascular arcade, at least 2 mm distal to the center of the fovea (Figure 7), avoiding direct contact with the retinal vasculature or with areas of pathologic features, such as dense atrophy or intraretinal pigment migration. Inject a small amount of the product slowly until an initial subretinal bleb is observed. Then inject the remaining volume slowly until the total 0.3 mL is delivered.
9. Following injection, discard all unused product. Dispose of the back-up syringe according to local biosafety guidelines applicable for handling and disposal of the product.
10. Perform a fluid-air exchange, carefully avoiding fluid drainage near the retinotomy created for the subretinal injection.
11. Initiate supine head positioning immediately in the post-operative period.
12. Upon discharge, advise patients to rest in a supine position as much as possible for 24 hours.
Special populations. Elderly (65 years or above). The safety and efficacy of voretigene neparvovec in patients ≥ 65 years old have not been established. However, no adjustment in dosage is necessary for elderly patients.
Hepatic and renal impairment. The safety and efficacy of voretigene neparvovec have not been established in patient with hepatic or renal impairment. No dose adjustment is required in these patients.
Paediatric population (below 18 years of age). The safety and efficacy of voretigene neparvovec in children below the age of 4 years have not been established. No dose adjustment is necessary for paediatric patients aged 4 years and above.
4.3 Contraindications
Hypersensitivity to the active substance(s) or to any of the excipients (see Section 6.1 List of Excipients).
Ocular or periocular infection.
Active intraocular inflammation.
4.4 Special Warnings and Precautions for Use
Endophthalmitis. Endophthalmitis may occur following any intraocular surgical procedure or injection. Use proper aseptic injection technique when administering Luxturna. Following the injection, monitor patients to permit early treatment of any infection. Advise patients to report any signs or symptoms of infection or inflammation without delay.
Patients should avoid swimming because of an increased risk of infection in the eye. Patients may resume swimming after a minimum of one to two weeks, on the advice of their healthcare professional.
Permanent decline in visual acuity. Permanent decline in visual acuity may occur following subretinal injection of Luxturna. Monitor patients for visual disturbances.
Retinal abnormalities. Retinal abnormalities may occur during or following the subretinal injection of Luxturna, including macular holes, foveal thinning, loss of foveal function, foveal dehiscence, and retinal haemorrhage. Monitor and manage these retinal abnormalities appropriately. Do not administer Luxturna in the immediate vicinity of the fovea (see Section 4.2 Dose and Method of Administration).
Retinal abnormalities may occur during or following vitrectomy including retinal tears, epiretinal membrane, or retinal detachment. Monitor patients during and following the injection to permit early treatment of these retinal abnormalities. Advise patients to report any signs or symptoms of retinal tears and/or detachment without delay.
Increased intraocular pressure. Increased intraocular pressure may occur after subretinal injection of Luxturna. Monitor and manage intraocular pressure appropriately.
Expansion of intraocular air bubbles. Instruct patients to avoid air travel or travel to high elevations until the air bubble formed following administration of Luxturna has completely dissipated from the eye. A time period of up to one week or more following injection may be required before dissipation of the air bubble. Verify the dissipation of the air bubble through ophthalmic examination. A rapid increase in altitude while the air bubble is still present can cause a rise in eye pressure and irreversible vision loss.
Vector shedding. Transient and low level vector shedding may occur in patient tears (see Section 5.2 Pharmacokinetic Properties, Clinical data).
As a precautionary measure, patients/caregivers should be advised to handle waste material generated from dressings, tears and nasal secretion appropriately, which may include storage of waste material in sealed bags prior to disposal. These handling precautions should be followed for 14 days after administration of Luxturna. It is recommended that patients/caregivers wear gloves for dressing changes and waste disposal, especially in case of underlying pregnancy, breastfeeding and immunodeficiency of caregivers.
Patients treated with Luxturna should not donate blood, organs, tissues and cells for transplantation.
Cataract. Subretinal injection of Luxturna, especially vitrectomy surgery, is associated with an increased incidence of cataract development and/or progression.
Use in hepatic impairment. See Section 4.2 Dose and Method of Administration, Special populations.
Use in renal impairment. See Section 4.2 Dose and Method of Administration, Special populations.
Use in the elderly. See Section 4.2 Dose and Method of Administration, Special populations.
Paediatric use. See Section 4.2 Dose and Method of Administration, Special populations.
Effects on laboratory tests. No data are available.
4.5 Interactions with Other Medicines and Other Forms of Interactions
No interaction studies have been performed.
4.6 Fertility, Pregnancy and Lactation
Considering the subretinal route of administration of Luxturna, and based on non-clinical and clinical data from trials of AAV2 vectors, there is a very low or negligible risk of inadvertent germ line transmission with AAV vectors.
Effects on fertility. No animal or clinical studies have been conducted to evaluate the effects of voretigene neparvovec on fertility.
Use in pregnancy. (Category B2)
There are no adequate and well-controlled studies in pregnant women to inform a product-associated risk.
Embryofetal development studies in animals have not been conducted with voretigene neparvovec.
As a precautionary measure, it is preferable to avoid the use of Luxturna during pregnancy.
Use in lactation. It is not known if voretigene neparvovec is present in human milk. There are no data on the effects of voretigene neparvovec on the breastfed infant or on milk production. A decision must be made whether to discontinue breastfeeding or to abstain from voretigene neparvovec therapy taking into account the benefit of breastfeeding for the child and the benefit of therapy for the mother.
4.7 Effects on Ability to Drive and Use Machines
Voretigene neparvovec has minor influence on the ability to drive and use machines. Patients may experience temporary visual disturbances after receiving subretinal injection of Luxturna. Patients should not drive or use heavy machines until visual function has recovered sufficiently, as advised by their ophthalmologist.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile. There were three non-serious adverse reactions of retinal deposits in three of 41 (7%) subjects that were considered to be related to voretigene neparvovec. All three of these events were a transient appearance of asymptomatic subretinal precipitates inferior to the retinal injection site, 1 to 6 days after injection and resolved without sequelae.
Serious adverse reactions related to the administration procedure were reported in three subjects during the clinical program. Increased intraocular pressure, which resulted in optic atrophy, was reported in one subject (1/41; 2%) secondary to administration of depo-steroid given to treat endophthalmitis related to the administration procedure. Retinal disorder (loss of foveal function) and retinal detachment were each reported in one subject each (1/41; 2%).
The most common ocular adverse reactions (incidence ≥ 5%) related to the administration procedure were conjunctival hyperaemia, cataract, increased intraocular pressure, retinal tear, dellen (thinning of the corneal stroma), macular hole, subretinal deposits, eye inflammation, eye irritation, eye pain, and maculopathy (wrinkling on the surface of the macula).
Tabulated summary of adverse drug reactions from clinical trials. The safety data described in this section reflect exposure to voretigene neparvovec in three clinical trials consisting of 41 subjects (81 eyes) with vision loss due to inherited retinal dystrophy caused by confirmed biallelic RPE65 mutation. Study 101 (n=12) was a Phase 1 safety and dose escalation study in which 12 subjects received unilateral subretinal injections of voretigene neparvovec. Eleven of the twelve subjects who participated in the dose escalation study went on to receive voretigene neparvovec in the second eye (Study 102). Study 301 (n=29) was an open-label, randomised, controlled study for both efficacy and safety (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
In total, 40 of the 41 subjects received sequential subretinal injections of voretigene neparvovec to each eye. One subject received voretigene neparvovec in only one eye. Seventy-two of the 81 eyes were exposed to the recommended dose of Luxturna at 1.5 x 1011 vg. In Study 101, 9 eyes were exposed to lower doses of voretigene neparvovec. The average age of the 41 subjects was 17 years ranging from 4 to 44 years. Of the 41 subjects, 25 (61%) were paediatric subjects under 18 years of age, and 23 (56%) were females.
Adverse drug reactions from clinical trials (Table 2) are listed by MedDRA system organ class. Within each system organ class, the adverse drug reactions are ranked by frequency, with the most frequent reactions first. Within each frequency grouping, adverse drug reactions are presented in order of decreasing seriousness. In addition, the corresponding frequency category for each adverse drug reaction is based on the following convention (CIOMS III): very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000).
Adverse reactions may have been related to voretigene neparvovec, the subretinal injection procedure, the concomitant use of corticosteroids, or a combination of these procedures and products.
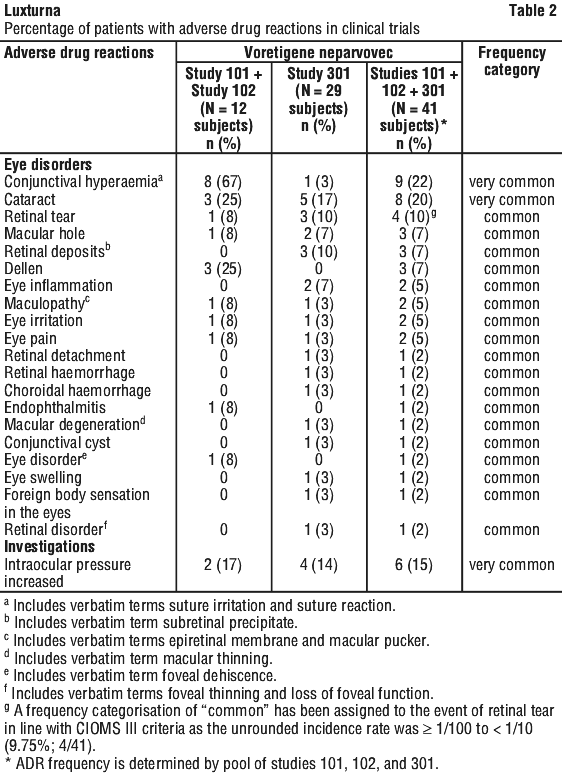
Eye disorders. Chorioretinal atrophy (includes preferred terms retinal degeneration, retinal depigmentation and injection site atrophy).
Description of selected adverse drug reactions. Chorioretinal atrophy. Chorioretinal atrophy has been reported as an adverse reaction during post-marketing experience and reported as progressive in some patients. Events were temporarily related to treatment and occurred in the estimated treated area of the bleb site and outside the bleb area. Retinal atrophy may involve the fovea with possible negative effects on the central vision.
Immunogenicity. At all doses of Luxturna evaluated in Studies 101 and 301, immune reactions were mild in severity and extra-ocular exposure was limited. In Study 101, the interval between the subretinal injections into the two eyes ranged from 1.7 to 4.6 years. In Study 301, the interval between the subretinal injections into the two eyes ranged from 7 to 14 days. No subject had a clinically significant cytotoxic T-cell response to either adeno-associated virus serotype 2 [AAV2] vector or retinal pigment epithelial 65 kDa protein [RPE65].
Subjects received systemic corticosteroids before and after subretinal injection of Luxturna to each eye. The corticosteroids may have decreased the potential immune reaction to either vector capsid [AAV2] or transgene product [RPE65].
Reporting of suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
4.9 Overdose
Symptomatic and supportive treatment is advised in case of overdose.
For information on the management of overdose, contact the Poisons Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: other ophthalmologicals SO1XA, ATC code: S01XA27.
Mechanism of action. Luxturna is designed to deliver a normal copy of the gene encoding the human retinal pigment epithelial 65 kDa protein (RPE65) to cells of the retina in persons with reduced or absent levels of biologically active RPE65. The RPE65 is produced in the retinal pigment epithelial (RPE) cells and converts all-trans-retinol to 11-cis-retinol, which subsequently forms the chromophore, 11 cis-retinal, during the visual (retinoid) cycle. The visual cycle is critical in phototransduction, which refers to the biological conversion of a photon of light into an electrical signal in the retina. Mutations in the RPE65 gene lead to reduced or absent levels of RPE65 isomerohydrolase activity, blocking the visual cycle, resulting in impairment of vision and ultimately complete blindness.
Pharmacodynamics (PD). Injection of Luxturna into the subretinal space results in transduction of some retinal pigment epithelial cells with a cDNA encoding normal human RPE65 protein, thus providing the potential to restore the visual cycle.
Clinical trials. Study 301. The efficacy of Luxturna in paediatric and adult patients was evaluated in an open-label, two-centre, randomised trial (Study 301).
Individuals with a confirmed genetic diagnosis of biallelic RPE65 gene mutations were eligible for enrolment if:
Both eyes had visual acuity of 20/60 (equivalent to 0.48 logMAR) or worse, and/or visual field less than 20 degrees in any meridian as measured by III4e isopter or equivalent;
They had sufficient viable retinal cells as determined by either: retinal thickness on spectral domain optical coherence tomography (> 100 microns within the posterior pole), at least 3 disc areas of the retina without atrophy or pigmentary degeneration within the posterior pole on ophthalmoscopy, or remaining visual field within 30 degrees of fixation as measured by III4e isopter or equivalent;
They were able to perform a standardised multi-luminance mobility test (MLMT) within the luminance range evaluated, but unable to pass the MLMT at 1 lux, the lowest luminance level tested.
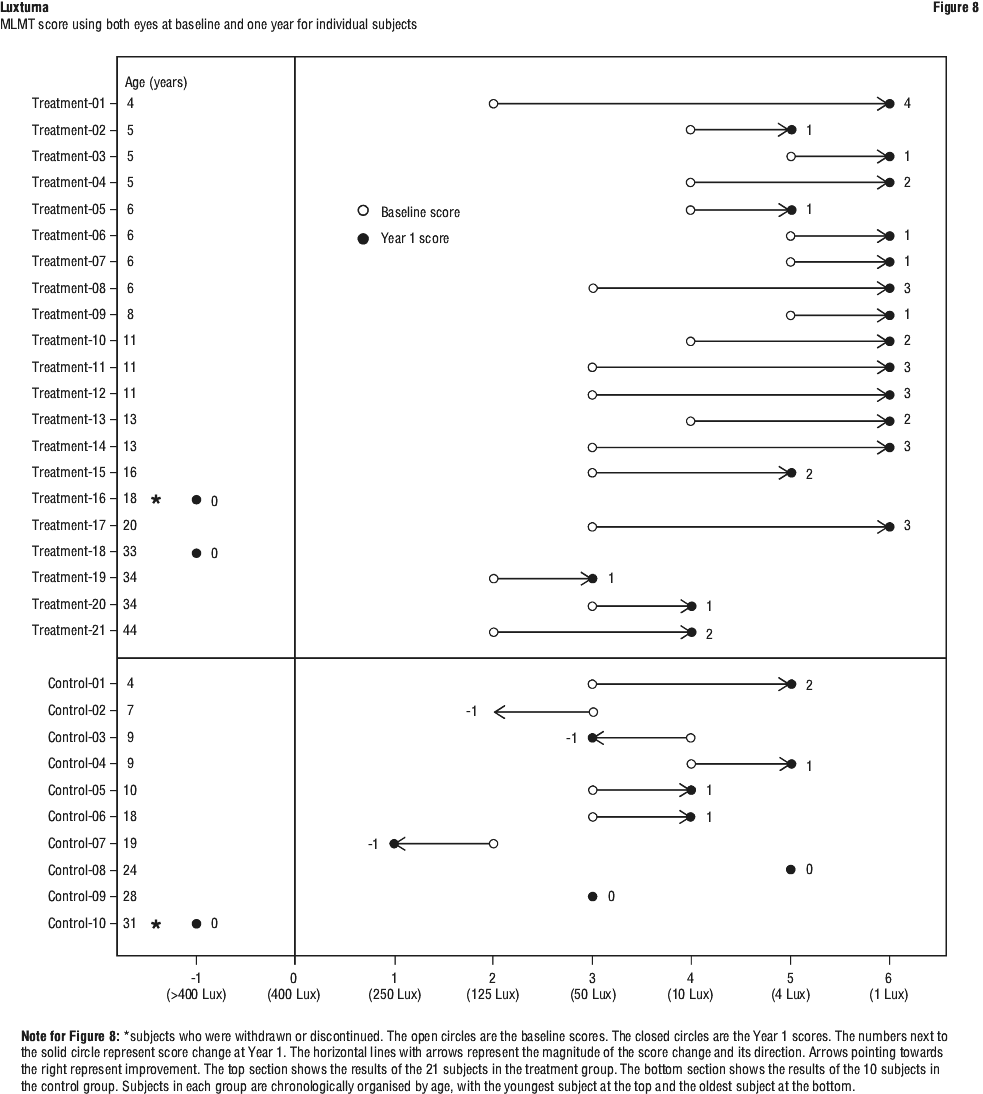
Of the 31 enrolled subjects, 21 subjects were randomised to receive subretinal injection of Luxturna. One subject discontinued from the study prior to treatment. Ten subjects were randomised to the control (non-intervention) group. One subject in the control group withdrew consent and was discontinued from the study. The nine subjects who were randomised to the control group were crossed over to receive subretinal injection of Luxturna after one year of observation. The average age of the 31 randomised subjects was 15 years (range 4 to 44 years), including 64% paediatric subjects (n=20, age from 4 to 17 years) and 36% adults (n=11). The average visual acuity at treatment baseline was 1.18 logMAR for the intervention group and 1.29 logMAR for the control. Bilateral subretinal injections of Luxturna were administered sequentially in two separate surgical procedures with an interval of 6 to 18 days.
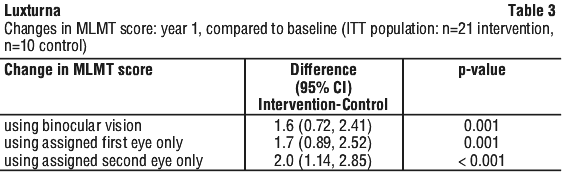
The efficacy of Luxturna was established on the basis of MLMT score change from Baseline to Year 1.
The MLMT was designed to measure changes in functional vision, as assessed by the ability of a subject to navigate a course accurately and at a reasonable pace at different levels of environmental illumination.
The MLMT was assessed using both eyes (binocular vision) and each eye separately at one or more of seven levels of illumination, ranging from 400 lux (corresponding to a brightly lit office) to 1 lux (corresponding to a moonless summer night). Each light level was assigned a score code ranging from 0 to 6. A higher score indicated that a subject was able to pass the MLMT at a lower light level. The MLMT of each subject was videotaped and assessed by independent graders using a defined combination of speed and accuracy scores. The MLMT score was determined by the lowest light level at which the subject was able to pass the MLMT. The MLMT score change was defined as the difference between the score at Baseline and the score at Year 1. A positive MLMT score change from Baseline to Year 1 visit indicated that the subject was able to complete the MLMT at a lower light level.
Three secondary endpoints were also tested: full-field light sensitivity threshold (FST) testing using white light; the change in MLMT score for the first assigned eye; and visual acuity (VA) testing.
Table 3 summarises the average MLMT score change from Baseline to Year 1 in the Luxturna treatment group compared to the control group.
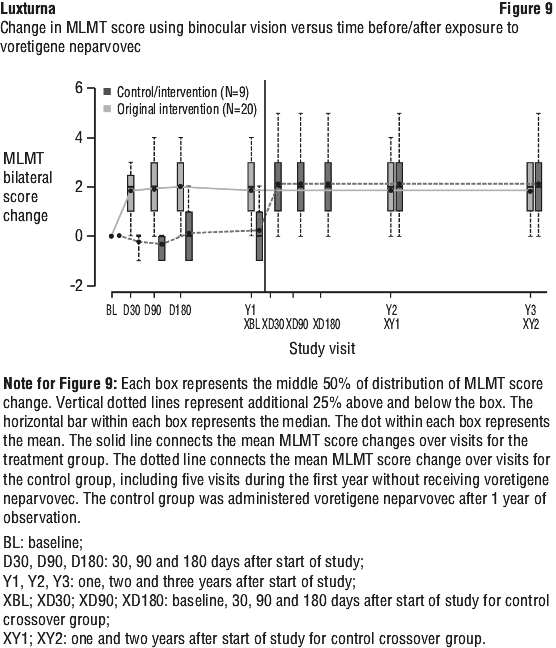
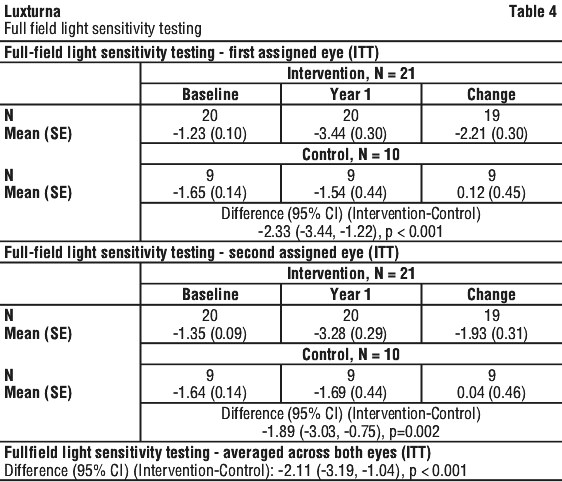
A supportive analysis showed that the linear relationships between the MLMT scores and FST in this study were generally good to strong, indicating that subjects with improvement on mobility testing at Year 1 tended to have lower (i.e. better) FST results at Year 1.
At one year after exposure to voretigene neparvovec, the mean change from baseline in visual acuity across both eyes using the Holladay scale was -0.16 LogMAR for the intervention group, and 0.01 LogMAR for the untreated control group. This reflected a mean 8 ETDRS-letter improvement for intervention subjects, compared to a mean 0.5-letter loss for control subjects. This difference between groups was not statistically significant.
In a supportive post hoc analysis using the Lange scale for off-chart scoring, the intervention group showed a 9.0 letter improvement versus a 1.5 letter improvement in the control group, averaged over both eyes (difference of 7.5 letters). This difference between groups was statistically significant.
5.2 Pharmacokinetic Properties
Pharmacokinetics (PK). Biodistribution (within the body) and vector shedding (excretion/secretion). Luxturna vector DNA levels in various tissues and secretions were determined using a quantitative polymerase chain reaction (qPCR) assay.
Nonclinical data. Biodistribution of voretigene neparvovec was evaluated at three months following subretinal administration in non-human primates. The highest levels of vector DNA sequences were detected in intraocular fluids (anterior chamber fluid and vitreous) of vector-injected eyes. Low levels of vector DNA sequences were detected in the optic nerve of the vector-injected eye, optic chiasm, spleen and liver, and sporadically in the lymph nodes. Vector DNA sequences were not detected in the gonads.
Clinical data. Luxturna vector shedding and biodistribution were investigated in a study measuring Luxturna DNA in tears from both eyes, and from serum, and whole blood of subjects in Study 301. In summary, Luxturna vector was shed transiently and at low levels in tears from the injected eye in 45% of the subjects in Study 301, and occasionally (7%) from the uninjected eye until Day 3 post-injection.
In 29 subjects who received bilateral administrations, Luxturna vector DNA was present in tear samples of 13 subjects (45%). Peak levels of vector DNA were detected in the tear samples on Day 1 post-injection, after which no vector DNA was detected in a majority of the subjects (8 of 13). Three subjects (10%) had vector DNA in tear samples until Day 3 postinjection, and one subject (3%) had vector DNA in tear samples until Day 14 post-injection. In another two subjects (7%), vector DNA was detected in tear samples from the uninjected (or previously injected) eye until Day 3 post-injection. Vector DNA was detected in serum in 3/29 (10%) subjects, including two with vector DNA in tear samples up to Day 3 following each injection.
5.3 Preclinical Safety Data
Bilateral, simultaneous subretinal administration of voretigene neparvovec was well tolerated at dose levels up to 8.25 x 1010 vg per eye in dogs with a naturally occurring RPE65 mutation and 7.5 x 1011 vg (5 times higher than the recommended human dose level) per eye in monkeys with normal-sighted eyes. In both animal models, bilateral, sequential subretinal administrations, where the contralateral eye was injected following the first eye, were well tolerated at the recommended human dose level of 1.5 x 1011 vg per eye. In addition, dogs with the RPE65 mutation displayed improved visual behaviour and pupillary responses.
Ocular histopathology of dog and non-human primate (monkey) eyes exposed to voretigene neparvovec showed only mild changes, which were mostly related to healing from surgical injury that expressed minimal RPE65 protein. In an earlier toxicology study, a similar AAV2 vector administered subretinally in dogs at a dose of 10 times the recommended dose resulted in focal retinal toxicity and inflammatory cell infiltrates histologically in regions exposed to the vector. Other findings in dogs and monkeys injected subretinally with voretigene neparvovec included occasional and isolated inflammatory cells in the retina, with no apparent retinal degeneration. Following a single vector administration, dogs developed antibodies to the AAV2 vector capsid whereas monkeys did not.
Genotoxicity. No genotoxicity studies have been conducted with voretigene neparvovec. The risk of insertional mutagenesis with voretigene neparvovec is considered to be low. Voretigene neparvovec lacks the AAV wildtype Rep proteins that catalyse site-specific viral genome integration.
Carcinogenicity. No animal carcinogenicity studies have been conducted with voretigene neparvovec.
6 Pharmaceutical Particulars
6.1 List of Excipients
Concentrate. Sodium chloride, monobasic sodium phosphate (for pH adjustment), dibasic sodium phosphate (for pH adjustment), poloxalene, water for injections.
Diluent. Sodium chloride, monobasic sodium phosphate (for pH adjustment), dibasic sodium phosphate (for pH adjustment), poloxalene, water for injections.
6.2 Incompatibilities
In the absence of compatibility studies, this product must not be mixed with other medicinal products.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Concentrate and diluent. Must be stored frozen at ≤ -65°C.
After thawing. Once thawed, the medicinal product should not be re-frozen and should be left at room temperature (below 25°C).
Following dilution under aseptic conditions. The solution must be used immediately; if not used immediately, the storage time at room temperature (below 25°C) should be no longer than 4 hours.
6.5 Nature and Contents of Container
0.5 mL extractable volume of concentrate in 2 mL cyclic olefin polymer vial with a chlorobutyl rubber stopper sealed in place with an aluminium flip-off seal.
1.7 mL extractable volume of solvent in a 2 mL cyclic olefin polymer vial with a chlorobutyl rubber stopper sealed in place with an aluminium flip-off seal.
Each foil pouch includes a carton containing 1 vial of concentrate and 2 vials of solvent.
6.6 Special Precautions for Disposal
This medicine contains genetically modified organisms. Unused medicine and waste products must be disposed of in compliance with the institutional guidelines for genetically modified organisms or clinical biohazardous waste, as appropriate.
6.7 Physicochemical Properties
CAS number. 1646819-03-5.
7 Medicine Schedule (Poisons Standard)
Not determined.
Date of First Approval
05 August 2020
Date of Revision
11 November 2025
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.