Mektovi
Brand Information
| Brand name | Mektovi |
| Active ingredient | Binimetinib |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Mektovi.
Summary CMI
MEKTOVI®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I using MEKTOVI?
MEKTOVI contains the active ingredient binimetinib. MEKTOVI can be used
- in combination with a medicine called encorafenib (BRAFTOVI®) to treat adult patients with a type of skin cancer called melanoma, which has spread to other parts of the body, or cannot be removed by surgery; or
- in combination with a medicine called encorafenib (BRAFTOVI®) to treat adult patients with a type of lung cancer called nonsmall cell lung cancer (NSCLC), when the cancer has spread to other parts of the body.
For more information, see Section 1. Why am I using MEKTOVI? in the full CMI.
2. What should I know before I use MEKTOVI?
Do not use if you have ever had an allergic reaction to or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use MEKTOVI? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with MEKTOVI and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I take MEKTOVI?
- The recommended dose of MEKTOVI, when taken in combination with BRAFTOVI, is 45 mg (three 15 mg tablets or one 45 mg tablet) twice daily taken about 12 hours apart (corresponding to a daily dose of 90 mg).
- Swallow the tablets whole with a full glass of water. MEKTOVI can be taken with or without food.
More instructions can be found in Section 4. How do I take MEKTOVI? in the full CMI.
5. What should I know while using MEKTOVI?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using MEKTOVI? in the full CMI.
6. Are there any side effects?
When MEKTOVI was taken with encorafenib, very common side effects included fatigue, nausea, diarrhoea, vomiting, problems with vision, stomach pain, joint pain, abnormal blood test results and muscle pain. For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
MEKTOVI®
Active ingredient(s): binimetinib
Consumer Medicine Information (CMI)
This leaflet provides important information about using MEKTOVI. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using MEKTOVI.
Where to find information in this leaflet:
1. Why am I using MEKTOVI?
2. What should I know before I use MEKTOVI?
3. What if I am taking other medicines?
4. How do I take MEKTOVI?
5. What should I know while using MEKTOVI?
6. Are there any side effects?
7. Product details
1. Why am I using MEKTOVI?
MEKTOVI contains the active ingredient binimetinib MEKTOVI is an anti-cancer medicine, which belongs to a group of medicines called ‘MEK inhibitors’.
MEKTOVI is used in combination with another medicine which contains the active ingredient, encorafenib (called BRAFTOVI®) to treat adult patients with a type of skin cancer called melanoma, which has spread to other parts of the body, or cannot be removed by surgery.
MEKTOVI is used in combination with another medicine which contains the active ingredient, encorafenib (called BRAFTOVI®) to treat adult patients with a type of lung cancer called non-small cell lung cancer (NSCLC) when the cancer which has spread to other parts of the body.
The type of cancers which MEKTOVI and BRAFTOVI are used to treat have a particular change (mutation) in a gene called BRAF. This mutation in the BRAF gene may have produced proteins which caused your cancer to develop.
Before you start treatment, your doctor will have tested you to confirm that you have this BRAF mutation.
MEKTOVI targets a protein called MEK which promotes cancer cell growth When MEKTOVI is used in combination with BRAFTOVI (which targets the changed BRAF protein), it further slows down or stops the growth of your cancer.
Ask your doctor if you have any questions about why this medicine has been prescribed for you.
Your doctor may have prescribed it for another reason.
BRAFTOVI is not recommended for children and adolescents aged under 18 years. The safety and efficacy of this medicine has not been established in this age group.
2. What should I know before I use MEKTOVI?
MEKTOVI is to be used in combination with BRAFTOVI, therefore you should also read the CMI for the other medicine you are planning to take.
Warnings
Do not use MEKTOVI if:
- you are allergic to binimetinib, or any of the ingredients listed at the end of this leaflet. Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin - Always check the ingredients to make sure you can use this medicine.
Check with your doctor if you:
- have any other medical conditions such as:
- heart problems
- high blood pressure
- muscle problems
- blood clots
- liver problems
- lung or breathing problems
- bleeding problems or if you are taking medicines that may increase your risk of bleeding
- eye problems including glaucoma or increased pressure in your eyes - take any medicines for any other condition
MEKTOVI contains lactose. Tell your doctor, nurse or pharmacist if you have an intolerance to some sugars.
Tell your doctor if you have had a history of blockage in the vein draining the eye (retinal vein occlusion) as MEKTOVI is not recommended in patients with a history of retinal vein occlusion.
Tell your doctor if you have had a different type of cancer than melanoma or NSCLC as MEKTOVI, when taken with BRAFTOVI may cause progression of certain other types of cancers.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
Taking MEKTOVI during pregnancy is not recommended. Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding before taking MEKTOVI.
MEKTOVI may cause permanent harm or birth defects to an unborn baby.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
MEKTOVI is not recommended while breast-feeding. If you are breastfeeding or planning to breastfeed, you must tell your doctor before taking this medicine.
It is not known if MEKTOVI passes into breastmilk.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interfere with MEKTOVI and affect how it works.
Keep a list of the medicines you take so you can show it to your doctor, nurse or pharmacist when you get a new medicine.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect MEKTOVI.
4. How do I take MEKTOVI?
How much to take
- Always take MEKTOVI exactly as your doctor has prescribed.
- The recommended dose of MEKTOVI, when taken in combination with BRAFTOVI, is 45 mg (three 15 mg tablets or one 45 mg tablet) twice daily taken about 12 hours apart (corresponding to a daily dose of 90 mg).
- If you experience serious side effects (such as skin, eye, heart or lung problems), your doctor may lower the dose of MEKTOVI, or stop treatment temporarily or permanently.
- Follow the instructions provided and use MEKTOVI until your doctor tells you to stop.
When to take MEKTOVI
- Swallow the tablets whole with a full glass of water.
- MEKTOVI can be taken with or without food.
- If vomiting occurs at any time after taking the tablets do not take an additional dose. Take the next dose as scheduled.
Continue taking MEKTOVI for as long as your doctor tells you to. Do not stop unless your doctor advises you to.
If you forget to use MEKTOVI
If you miss a dose of MEKTOVI at the usual time, the missed dose is less than 6 hours late, take it as soon as you remember.
If the missed dose is more than 6 hours late, skip that dose and take your next dose at the usual time.
Then go back to taking your tablets as you would normally
Do not take a double dose to make up for the dose that you missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your doctor, nurse or pharmacist for some hints.
If you use too much MEKTOVI
If you think that you have used too much MEKTOVI, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using MEKTOVI?
Things you should do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking MEKTOVI.
If you are going to have surgery, tell the surgeon or that you are taking MEKTOVI.
If you become pregnant while taking MEKTOVI, tell your doctor immediately.
If you are a woman who could become pregnant, you must use effective birth control (contraception) while you are taking MEKTOVI, and you must continue to use effective contraception for at least 1 month after taking your last dose.
Tell your doctor if you are breastfeeding while being treated with MEKTOVI.
Call your doctor straight away if you experience the following while you are taking MEKTOVI:
- Skin changes
MEKTOVI when taken with BRAFTOVI may cause other types of skin cancer such as cutaneous squamous cell carcinoma.
Your doctor will periodically check for new cancers on your skin and inside your body before, during and after your treatment. Tell your doctor immediately if you detect any skin changes including new warts, skin soreness, reddish bumps which bleed or don't heal or any changes in the size or colour of a mole. - Heart problems
MEKTOVI can lower the amount of blood pumped by your heart or make existing heart problems worse. Your doctor will run tests to check that your heart is working properly before and during your treatment with this medicine. - Blood clots
MEKTOVI can cause blood clots in your arms or legs which can travel to your lungs and lead to death. If necessary, your doctor may decide to interrupt or completely stop your MEKTOVI treatment. - Bleeding problems
MEKTOVI may cause serious bleeding problems. Tell your doctor immediately if you have any signs of bleeding. - Eye problems
MEKTOVI I may cause serious eye problems. Your doctor will examine your eyes for any new or worsening problems with your sight while you are taking these medicines. - Liver problems
MEKTOVI may increase the amounts of liver enzymes in your blood. Your doctor will run blood tests to monitor your liver function before and during treatment. - Muscle problems
MEKTOVI can cause breakdown of muscle (rhabdomyolysis). Your doctor will run blood tests to monitor muscle condition before and during treatment. As a precaution, drink plenty of fluids during treatment, unless otherwise advised by your doctor. - High blood pressure
MEKTOVI can raise blood pressure. Your doctor or nurse will check your blood pressure before and during treatment with MEKTOVI. - Lung or breathing problems
MEKTOVI may cause lung or breathing problems including inflammation of the lungs (pneumonitis or interstitial lung disease). Signs and symptoms can include: cough, shortness of breath or fatigue. If necessary, your doctor may interrupt or completely stop your MEKTOVI treatment.
If you experience the following symptoms, contact your doctor immediately as this can be a life-threatening condition: nausea, shortness of breath, irregular heartbeat, muscular cramps, seizures, clouding of urine, decrease in urine output and tiredness. These may be caused by a group of metabolic complications that can occur during treatment of cancer that are caused by the breakdown products of dying cancer cells (Tumour lysis syndrome (TLS)) and can lead to changes in kidney function (see also Section 6: Possible side effects).
Remind any doctor, dentist or pharmacist you visit that you are using MEKTOVI. Keep all of your doctor's appointments so that your progress can be checked.
Things you should not do
- Do not take MEKTOVI to treat any other complaints unless your doctor tells you to.
- Do not give your medicine to anyone else, even if their symptoms seem similar to yours or they have the same condition as you.
- Do not stop taking your medicine or lower the dosage without checking with your doctor
Driving or using machines
Be careful before you drive or use any machines or tools until you know how MEKTOVI affects you.
MEKTOVI can affect your ability to drive or use machines.
If you experience any problems with your vision, or any other side-effects that may affect your ability, avoid driving or using machines. Talk to your doctor if you are not sure if you should drive.
Looking after your medicine
- Keep your MEKTOVI capsules in their original pack until it is time to take them.
- Keep your MEKTOVI capsules in a place where the temperature stays below 30°C.
Follow the instructions in the carton on how to take care of your medicine properly.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Keep it where young children cannot reach it.
Getting rid of any unwanted medicine
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Do not throw any medicines away via wastewater or household waste.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
When MEKTOVI was taken with BRAFTOVI the following side effects were reported.
| Less serious side effects | What to do |
Head and neurology related:
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
| Heart related MEKTOVI can affect how well your heart pumps (left ventricular dysfunction). Signs and symptoms can include:
MEKTOVI can increase blood pressure. Tell your doctor immediately if you experience severe headache, feel dizzy or lightheaded, or if your blood pressure is much higher than usual (if you are self-monitoring your blood pressure at home). Allergy related: allergic reaction that may include swelling of the face and difficulty breathing Blood clot related MEKTOVI may cause blood clots (venous thromboembolism including pulmonary embolism). Signs and symptoms can include:
MEKTOVI may induce fluid leakage under the retina in the eye that results in detachment of different layers in the eye (retinal pigment epithelial detachment), which could lead to the following symptoms:
MEKTOVI may lead to breakdown of muscles (rhabdomyolysis) which can lead to kidney damage and can be fatal. Signs and symptoms can include:
Taking MEKTOVI can cause serious bleeding problems. Tell your doctor immediately if you have any unusual bleeding or signs of bleeding including:
MEKTOVI when taken with BRAFTOVI may cause other types of skin cancer Tumour lysis syndrome MEKTOVI can cause a rapid breakdown of cancer cells which in some people may be fatal. Symptoms may include nausea, shortness of breath, irregular heartbeat, muscular cramps, seizures, clouding of urine, decrease in urine output and tiredness. | Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What MEKTOVI contains
| Active ingredient (main ingredient) | 15 or 45 mg of binimetinib |
| Other ingredients (inactive ingredients) | Tablet core:
|
| Potential allergens | Lactose monohydrate |
Do not take this medicine if you are allergic to any of these ingredients.
What MEKTOVI looks like
MEKTOVI 15 mg tablets are supplied in blister packs of 84 tablets (7 strips of 12 tablets each).
The 15 mg tablets are yellow/dark yellow, unscored biconvex, oval and film-coated, with “A” debossed on one face and “15” on the opposite face.
15 mg: AUST R 295440
MEKTOVI 45 mg tablets are supplied in blister packs of 28 tablets (each blister contains 14 tablets each).
The 45 mg tablets are white to off-white, unscored biconvex, oval and film-coated, with "45" debossed on one face.
45 mg: AUST R 484525
Who distributes MEKTOVI
Pierre Fabre Australia Pty Limited
Level 7, 32 Walker St
North Sydney, NSW 2060
Australia
® = Registered Trademark
This leaflet was prepared in December 2025
Brand Information
| Brand name | Mektovi |
| Active ingredient | Binimetinib |
| Schedule | S4 |
MIMS Revision Date: 01 February 2026
1 Name of Medicine
Binimetinib.
2 Qualitative and Quantitative Composition
Each Mektovi 15 mg film-coated tablet contains binimetinib 15 mg.
Each Mektovi 45 mg film-coated tablet contains binimetinib 45 mg.
Contains lactose. Contains sugars. For a full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Mektovi 15 mg film-coated tablets are yellow/dark yellow, unscored biconvex, ovaloid film-coated tablets, approximately 12 mm in length and 5 mm in width, with the "A" logo debossed on one face of the tablet and "15" on the opposing face.
Mektovi 45 mg film-coated tablets are white to off-white, unscored biconvex, ovaloid film-coated tablets approximately 15 mm in length and 6 mm in width, with "45" debossed on one side.
4 Clinical Particulars
4.1 Therapeutic Indications
Melanoma. Binimetinib in combination with encorafenib is indicated for the treatment of adult patients with unresectable or metastatic melanoma with a BRAF V600E or V600K mutation, as detected by a validated test.
Non-small cell lung cancer (NSCLC). Binimetinib in combination with encorafenib is indicated for the treatment of adult patients with metastatic non-small cell lung cancer with a BRAF V600E mutation.
4.2 Dose and Method of Administration
Treatment with binimetinib in combination with encorafenib should only be initiated and supervised by a physician experienced in the use of anti-cancer medicines.
BRAF mutation testing. Prior to treatment with binimetinib in combination with encorafenib, the BRAF V600 mutation status of a patient's melanoma or NSCLC must be confirmed by a validated test, conducted by an experienced laboratory (see Section 5.1 Pharmacodynamic Properties, Clinical trials).The efficacy and safety of binimetinib in combination with encorafenib have been established only in patients with melanoma tumours expressing BRAF V600E and V600K mutations or NSCLC expressing a BRAF V600E mutation. Binimetinib in combination with encorafenib should not be used in patients with wild type BRAF malignant melanoma or wild-type BRAF NSCLC.
Dosage.The recommended dose of binimetinib is 45 mg (three 15 mg tablets or one 45 mg tablet) twice daily (corresponding to a total dose of 90 mg), approximately 12 hours apart, when used in combination with encorafenib.
Administration. Binimetinib tablets should be swallowed whole with water, with or without food.
Duration of treatment. Treatment should continue until the patient no longer derives benefit or unacceptable toxicity develops.
Missed dose. If a dose of binimetinib is missed, it should not be taken if it is less than 6 hours until the next dose is due.
Vomiting after administration. If a patient vomits after administration of binimetinib, the patient should not take the dose again. The patient should take the next scheduled dose.
Dose modification. The management of adverse reactions may require dose reduction, temporary interruption or treatment discontinuation (see below and Table 1). The decision on whether to modify the dose of binimetinib should be based on the prescriber's assessment of individual patient safety and tolerance.
The recommended reduced dose of binimetinib is 30 mg twice daily. Dose reduction below 30 mg twice daily is not recommended. Therapy should be discontinued if the patient is not able to tolerate 30 mg orally twice daily.
If the adverse reaction that resulted in a dose reduction is under effective management, re-escalation to 45 mg twice daily may be considered. Dose re-escalation to 45 mg twice daily is not recommended if the dose reduction is due to left ventricular dysfunction (LVD) or any Grade 4 toxicity.
If treatment-related toxicities occur when binimetinib is used in combination with encorafenib, then both treatments should be simultaneously dose reduced, interrupted or discontinued. Exceptions where dose modifications are necessary for encorafenib only (adverse reactions primarily related to encorafenib) are: palmar-plantar erythrodysaesthesia syndrome (PPES), uveitis including iritis and iridocyclitis, and QTc prolongation.
If one of these toxicities occurs, see Section 4.2. Dose and Method of Administration of encorafenib PI for dose modification instructions for encorafenib.
If binimetinib is temporarily interrupted, reduce encorafenib to 300 mg once daily during the time of binimetinib dose interruption (see Table 1) as encorafenib is not well-tolerated at the dose of 450 mg as a single agent. If binimetinib is permanently discontinued, encorafenib may be continued (at the reduced dose of 300 mg) depending on the individual clinical benefit.
If encorafenib is temporarily interrupted (see Section 4.2 Dose and Method of Administration of encorafenib PI), interrupt binimetinib. If encorafenib is permanently discontinued, then discontinue binimetinib.
Dose modification recommendations in case of adverse reactions are presented in Table 1. For information on the dosage and recommended dose modifications of encorafenib, refer to the encorafenib PI, see Section 4.2 Dose and Method of Administration.
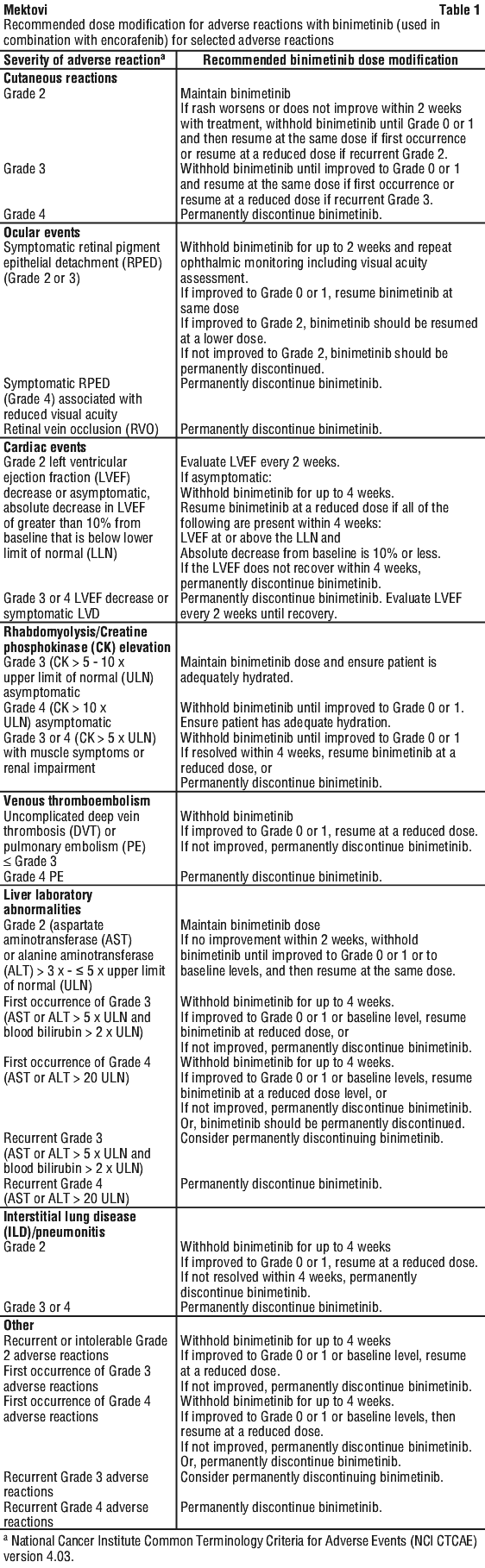
Renal impairment. No dose adjustment is required for patients with renal impairment (see Section 5.2 Pharmacokinetic Properties).
Elderly patients (65 years and older). No dose adjustment is required for elderly patients (see Section 5.2 Pharmacokinetic Properties).
Children and adolescents (< 18 years). The safety and efficacy of binimetinib have not been established in patients below the age of 18 years. There are no data available.
4.3 Contraindications
Hypersensitivity to the active substance binimetinib or to any of the excipients (see Section 6.1 List of Excipients).
4.4 Special Warnings and Precautions for Use
When binimetinib is given in combination with encorafenib, the PI for encorafenib must be consulted prior to initiation of combination treatment. For additional information on warnings and precautions associated with encorafenib treatment, please refer to the PI for encorafenib.
Binimetinib in combination with encorafenib in patients who have progressed on a BRAF inhibitor. There are limited data on the use of the combination of binimetinib with encorafenib in patients who previously progressed on a prior BRAF inhibitor treatment for unresectable or metastatic melanoma with a BRAF V600 mutation. These data show that the efficacy of the combination would be lower in these patients.
Binimetinib in combination with encorafenib in patients with brain metastases. There are limited efficacy data on the use of the combination of binimetinib and encorafenib in patients with a BRAF V600 mutant melanoma or BRAF V600E mutant NSCLC with brain metastases (see Section 5.1 Pharmacodynamic Properties).
Left ventricular dysfunction. Left ventricular dysfunction, defined as symptomatic or asymptomatic decreases in ejection fraction can occur with the use of binimetinib.
It is recommended that LVEF is assessed by echocardiogram or multi-gated acquisition (MUGA) scan before initiation of binimetinib, 1 month after initiation and then at approximately 3-month intervals or more frequently as clinically indicated while on treatment. The occurrence of LVEF decrease can be managed with dose reduction, treatment interruption or treatment discontinuation (see Section 4.2 Dose and Method of Administration).
The safety of binimetinib in combination with encorafenib has not been established in patients with a baseline LVEF that is either below 50% or below the institutional LLN. Therefore, in these patients, binimetinib should be used with caution and for any symptomatic LVD, Grade 3 or 4 LVEF, or absolute decrease of LVEF from baseline of ≥ 10%, binimetinib should be discontinued and LVEF should be evaluated every 2 weeks until recovery.
Venous thromboembolism. Venous thromboembolism (VTE) can occur with the use of binimetinib (see Section 4.8 Adverse Effects (Undesirable Effects)). Binimetinib should be used with caution in patients who are at risk of, or with a history of VTE.
If during treatment the patient develops VTE or pulmonary embolism, it should be managed with dose reduction, treatment interruption or treatment discontinuation (see Section 4.2 Dose and Method of Administration).
Haemorrhage. Haemorrhages, including major haemorrhagic events, can occur when binimetinib is administered with encorafenib (see Section 4.8 Adverse Effects (Undesirable Effects)). The risk of haemorrhage may be increased with concomitant use of anticoagulant and antiplatelet therapy. The occurrence of Grade ≥ 3 haemorrhagic events should be managed with dose reduction, treatment or treatment discontinuation; as clinically indicated (see Section 4.2 Dose and Method of Administration).
Ocular toxicities. Ocular toxicities including RPED and RVO can occur when binimetinib is administered. Uveitis, including iridocyclitis and iritis, was reported in patients treated with binimetinib in combination with encorafenib (see Section 4.8 Adverse Effects (Undesirable Effects)).
Binimetinib is not recommended in patients with a history of RVO. The safety of binimetinib has not been established in patients with predisposing factors for RVO including uncontrolled glaucoma, ocular hypertension, uncontrolled diabetes mellitus or a history of hyperviscosity or hypercoagulability syndromes. Binimetinib should therefore be used with caution in these patients.
Patients should be assessed at each visit for symptoms of new or worsening visual disturbances. If symptoms of new or worsening visual disturbances including diminished central vision, blurred vision or loss of vision are identified, a prompt ophthalmologic examination is recommended.
The occurrence of symptomatic RPED can be managed with dose reduction, treatment interruption or treatment discontinuation (see Section 4.2 Dose and Method of Administration, Table 1).
Binimetinib should be permanently discontinued with the occurrence of RVO (see Section 4.2 Dose and Method of Administration, Table 1).
If a patient develops uveitis during treatment, see Section 4.2 Dose and Method of Administration of encorafenib PI for guidance.
CK elevation and rhabdomyolysis. Asymptomatic CK elevations are seen in patients treated with binimetinib in combination with encorafenib (see Section 4.8 Adverse Effects (Undesirable Effects)). Across clinical trials of binimetinib in combination with encorafenib, rhabdomyolysis was uncommonly reported. Special attention should be paid to the use of binimetinib in patients with neuromuscular conditions associated with CK elevation and rhabdomyolysis.
CK and creatinine levels should be monitored monthly during the first 6 months of treatment and as clinically indicated. The patient should be advised to maintain an adequate fluid intake during treatment. Depending on the severity of symptoms, degree of CK elevation or creatinine elevation, dose reduction, dose interruption or permanent discontinuation of binimetinib may be required (see Section 4.2 Dose and Method of Administration).
New primary malignancies. New primary malignancies, cutaneous and non-cutaneous, have been observed in patients treated with BRAF inhibitors and can occur when binimetinib is administered in combination with encorafenib.
Cutaneous malignancies. Cutaneous malignancies such as cutaneous squamous cell carcinoma (cuSCC) including kerathoacanthoma has been observed in patients treated with binimetinib when used in combination with encorafenib.
Dermatologic evaluations should be performed prior to initiation of therapy with binimetinib in combination with encorafenib every 2 months while on therapy and for up to 6 months following discontinuation of the combination. Suspicious skin lesions should be managed with dermatological excision and dermatopathologic evaluation. Patients should be instructed to immediately inform their physicians if new skin lesions develop. Encorafenib and binimetinib should be continued without any dose modifications.
Non-cutaneous malignancies. Based on its mechanism of action, encorafenib may promote malignancies associated with activation of RAS through mutation or other mechanisms. Patients receiving binimetinib in combination with encorafenib should undergo a head and neck examination, chest/abdomen computerised tomography scan, anal and pelvic examinations (for women) and complete blood cell counts prior to initiation, during and at the end of treatment as clinically appropriate. Permanent discontinuation of binimetinib and encorafenib should be considered in patients who develop RAS mutation-positive non-cutaneous malignancies. Benefits and risks should be carefully considered before administering binimetinib in combination with encorafenib to patients with a prior or concurrent cancer associated with RAS mutation.
Hypertension. Hypertension, or worsening of pre-existing hypertension, can occur with the use of binimetinib. Blood pressure should be measured at baseline and monitored during treatment, with control of hypertension by standard therapy as appropriate. In case of severe hypertension, temporary interruption of binimetinib is recommended until hypertension is controlled (see Section 4.2 Dose and Method of Administration, Table 1).
Pneumonitis/interstitial lung disease (ILD). Pneumonitis/interstitial lung disease (ILD) can occur with binimetinib. Treatment with binimetinib should be withheld in patients with suspected pneumonitis or ILD, including patients presenting new or progressive pulmonary symptoms or findings such as cough, dyspnoea, hypoxia, reticular opacities or pulmonary infiltrates (see Section 4.2 Dose and Method of Administration, Table 1). Binimetinib should be permanently discontinued in patients diagnosed with treatment related pneumonitis or ILD.
Tumour lysis syndrome (TLS). The occurrence of TLS, which may be fatal, has been associated with the use of binimetinib in association with encorafenib (see Section 4.8 Adverse Effects (Undesirable Effects)). Risk factors for TLS include high tumour burden, pre-existing chronic renal insufficiency, oliguria, dehydration, hypotension and acidic urine. These patients should be monitored closely and treated promptly as clinically indicated, and prophylactic hydration should be considered.
Use in hepatic impairment. Liver metabolism mainly via glucuronidation is the primary route of elimination of binimetinib (see Section 5.2 Pharmacokinetic Properties). As encorafenib is not recommended in patients with moderate (Child Pugh B) and severe hepatic impairment (Child Pugh C), administration of binimetinib is not recommended in these patients (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties).
Lactose intolerance. Mektovi contains lactose. Patients with rare hereditary problems of galactose intolerance, total lactase deficiency or glucose-galactose malabsorption should not take Mektovi.
Use in the elderly. Please see Section 5.2 Pharmacokinetic Properties; Section 4.8 Adverse Effects (Undesirable Effects).
Paediatric use. The safety and efficacy of binimetinib in children and adolescents aged < 18 years have not yet been established. There are no data available.
Effects on laboratory tests. Liver laboratory abnormalities (AST, ALT elevations) can occur with binimetinib (see Section 4.8 Adverse Effects (Undesirable Effects)). Liver laboratory values should be monitored before initiation of binimetinib and encorafenib and at least monthly during the first 6 months of treatment and then as clinically indicated. Liver function abnormalities should be managed with dose reduction, treatment interruption or treatment discontinuation (see Section 4.2 Dose and Method of Administration).
4.5 Interactions with Other Medicines and Other Forms of Interactions
Effect of UGT1A1 and UGT2B7 inducers or inhibitors on binimetinib. Binimetinib is primarily metabolised by UGT1A1 and UGT2B7 mediated glucuronidation and to a lesser extent by CYP1A2- and CYP2C19-mediated oxidation. In a clinical study subanalysis however, there was no apparent relationship observed between binimetinib exposure and UGT1A1 mutation status. In addition, simulations to investigate the effect of 400 mg atazanavir (UGT1A1 inhibitor) on the exposure of 45 mg binimetinib predicted similar binimetinib Cmax in the presence or absence of atazanavir. Since binimetinib is metabolised by multiple enzymes, the possible extent of drug interactions mediated by UGT1A1, UGT2B7, CYP1A2 or CYP2C19 is minimal and unlikely to be clinically relevant; however, as this has not been evaluated in a formal clinical study, UGT1A1 or UGT2B7 inducers (such as rifampicin and phenobarbital), UGT1A1 inhibitors (such as indinavir, atazanavir and sorafenib) and UGT2B7 inhibitors (quinidine, mefenamic acid and diclofenac) should be coadministered with caution.
Combination with encorafenib. While encorafenib is a relatively potent reversible inhibitor of UGT1A1, no differences in binimetinib exposure have been observed clinically when binimetinib is co-administered with encorafenib.
Effect of transporters on binimetinib. In vitro experiments indicate that binimetinib is a substrate of P-glycoprotein (P-gp) and breast cancer resistance protein. Co-administration of binimetinib with inhibitors of these transporters may increase the plasma binimetinib concentration in patients.
No clinically relevant drug interactions have been demonstrated with binimetinib.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. There are no data on the effect of binimetinib on fertility in humans.
Fertility studies were not conducted with binimetinib. In repeat-dose toxicity studies, no concern on reproductive organs were observed in rats or monkeys (animal: human exposure ratios up to 19 and 0.4, respectively). It is uncertain whether binimetinib may affect fertility in patients.
Women of childbearing potential. Women of childbearing potential should be advised to use effective contraception during treatment with binimetinib and for at least 1 month after the last dose. Women of childbearing potential receiving binimetinib in combination with encorafenib should be advised that encorafenib may decrease efficacy of hormonal contraceptives. Therefore, female patients using hormonal contraception are advised to use an additional or alternative method such as a barrier method (e.g. condom) during treatment with encorafenib and for at least 1 month following the last dose.
Use in pregnancy. (Category D)
There are no data on the use of binimetinib in pregnant women. However, studies in animals have demonstrated reproductive toxicity. The potential embryo-foetal effects of binimetinib were evaluated in rats and rabbits. In rats, lower gestational body weight gain and foetal body weight were noted at ≥ 30 mg/kg/d and a decreased number of ossified foetal sternebrae was noted at ≥ 10 mg/kg/d (8 times the clinical exposure). The NOAEL in rats was 10 mg/kg/d. In rabbits, mortality, maternal physical signs of toxicity, lower gestational body weight and abortion were noted at ≥ 10 mg/kg/d (1.4 times the clinical exposure). From 10 mg/kg/d, the number of viable foetuses and foetal body weights were reduced and postimplantation loss and resorptions were increased. At 20 mg/kg/d, increased litter incidences of foetal ventricular septal defects, dilated aortic arch and pulmonary trunk alterations were noted. The NOAEL in rabbit was 2 mg/kg/d (0.5 times the clinical exposure).
If administered to pregnant women, binimetinib may harm the foetus. Binimetinib should not be administered during pregnancy unless the benefits for the mother clearly outweigh the risks for the foetus.
Use in lactation. It is not known if binimetinib or its active metabolite is excreted in human milk. Because many drugs are excreted in breast milk and because of the potential for serious adverse reactions in nursing infants, a decision should be made whether to discontinue binimetinib or to discontinue nursing, taking into account the benefit of breast feeding for the child and the benefit of the drug to the mother.
4.7 Effects on Ability to Drive and Use Machines
Visual disturbances have been reported in patients treated with binimetinib during clinical trials. Patients should be advised not to drive or use machines if they experience visual disturbances or any other adverse effects that may affect their ability to drive or use machines (see Section 4.8 Adverse Effects (Undesirable Effects)).
4.8 Adverse Effects (Undesirable Effects)
Summary of safety profile. Melanoma studies. The safety of binimetinib (45 mg orally twice daily) in combination with encorafenib (450 mg orally once daily) (hereafter referred to as the pooled Combo 450 population) was evaluated in 274 patients with BRAF V600 mutation-positive unresectable or metastatic melanoma, based on two Phase II studies (CMEK162X2110 and CLGX818X2109) and one Phase III study (CMEK162B2301).
At the recommended Combo 450 dose in patients with metastatic melanoma (n=274), the most common adverse reactions (> 25%) occurring in patients treated with binimetinib in combination with encorafenib were fatigue, nausea, diarrhoea, vomiting, retinal detachment, abdominal pain, arthralgia, blood CK increased and myalgia.
The safety of encorafenib (300 mg orally once daily) in combination with binimetinib (45 mg orally twice daily) was evaluated in 257 patients with BRAF V600 mutant unresectable or metastatic melanoma (hereafter referred to as the Combo 300 population), based on the Phase III study (CMEK162B2301, Part 2). The most common adverse reactions (≥ 25%) occurring in patients treated with encorafenib 300 mg administered with binimetinib were fatigue, nausea and diarrhoea.
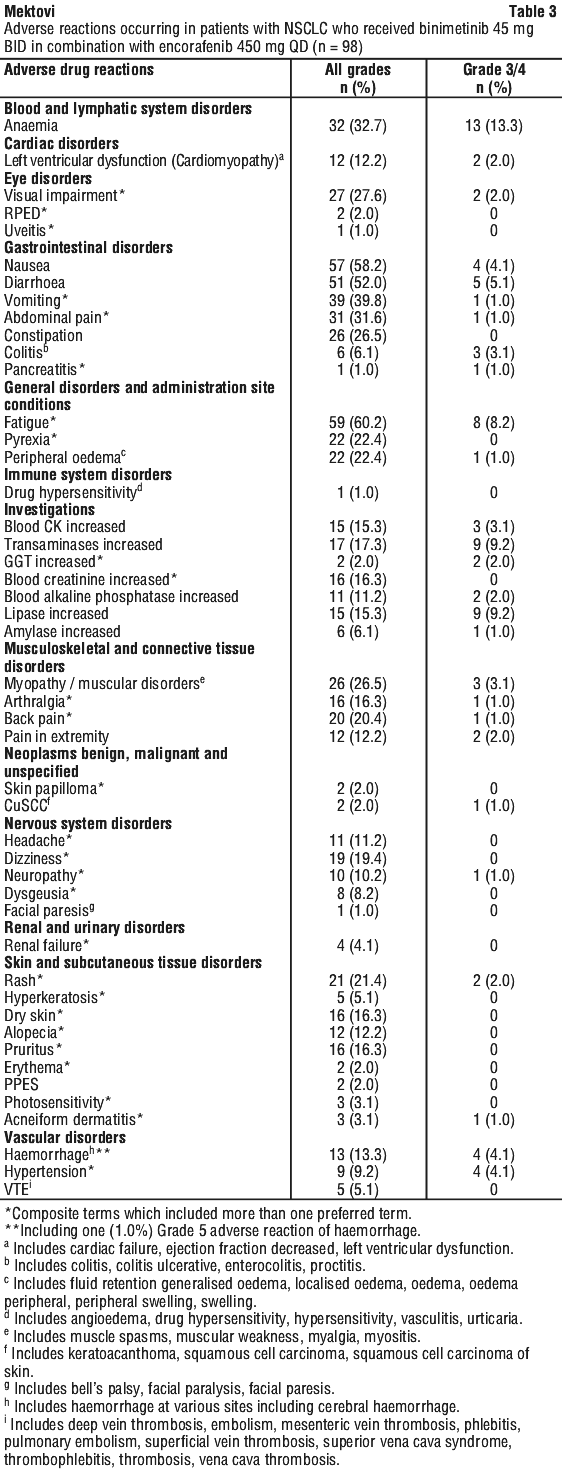
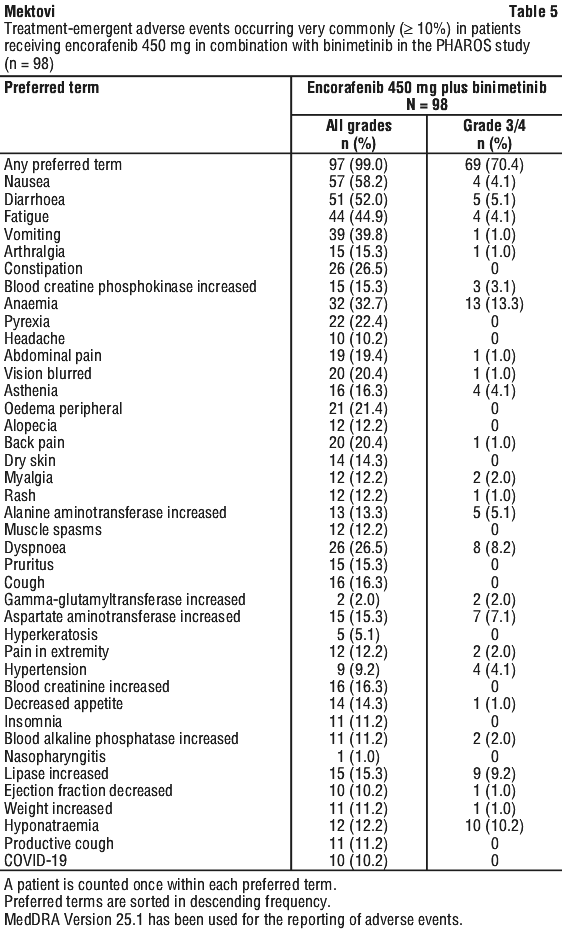
Non-small cell lung cancer studies. The safety of binimetinib (45 mg orally twice daily [BID]) in combination with encorafenib (450 mg orally once daily [QD]) was evaluated in 98 patients with BRAF V600E mutant metastatic Non-small cell lung cancer (NSCLC) who received this regimen in the open-label, single-arm trial ARRAY-818-202 "PHAROS" (see Section 5.1 Pharmacodynamic Properties, Clinical trials). The most common ADRs (> 25%) reported in this population were: fatigue, nausea, diarrhoea, vomiting, anaemia, abdominal pain, visual impairment, myopathy/muscular disorders and constipation.
Tabulated summary of adverse reactions. Melanoma studies. Adverse reactions in the pooled Combo 450 population (n=274) are listed in Table 2 by MedDRA body system organ class (SOC).
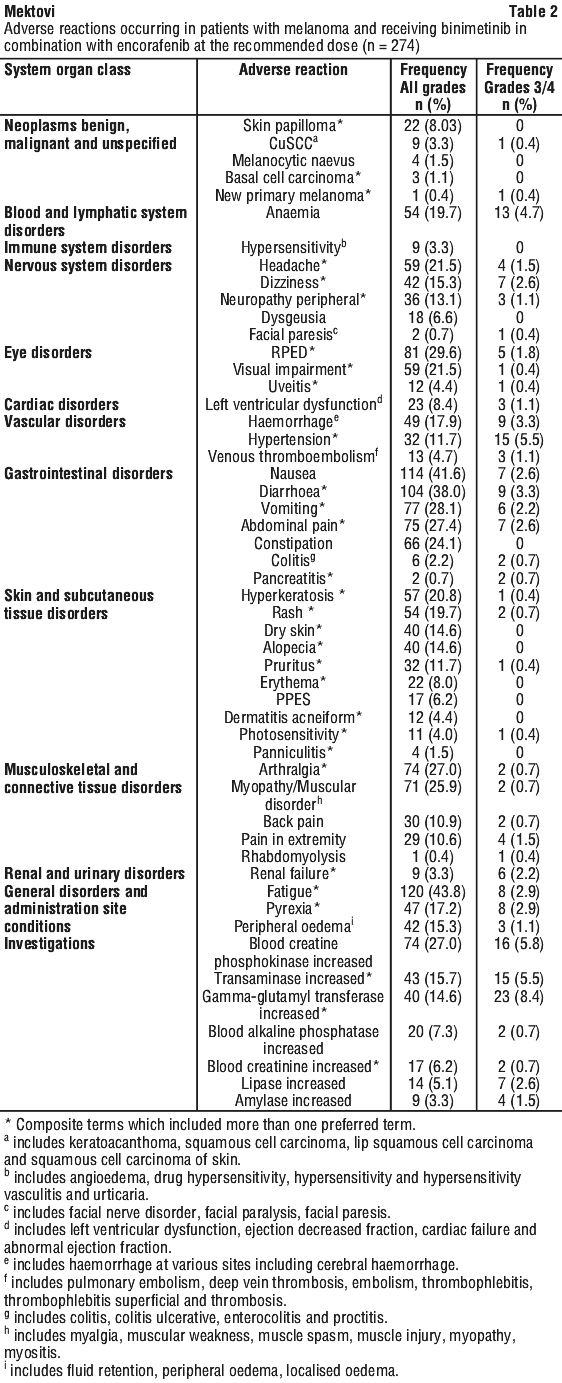
Ocular events. Melanoma studies. In the pooled Combo 450 population, RPED was reported in 29.6% (81/274) of patients. RPED was Grade 1 (asymptomatic) in 21.2% (58/274) of patients, Grade 2 in 6.6% (18/274) and Grade 3 in 1.8% (5/274). Most of these events were reported as retinopathy (9.5%, 26/274), retinal detachment (6.6%, 18/274), subretinal fluid (6.2%, 17/274), macular oedema (5.1%, 14/274) and chorioretinopathy (3.3%, 9/274), and led to dose interruptions or dose modifications in 4.7% (13/274) of patients. The median time to onset of the first event of RPED (all grades) was 1.5 month (range 0.03 to 17.5 months). RPED was generally reversible. Visual impairment, including vision blurred and reduced visual acuity, occurred in 21.5% (59/274) of patients. Visual impairment was generally reversible.
Uveitis was reported when binimetinib was used in combination with encorafenib (see Section 4.8 Adverse Effects (Undesirable Effects) of encorafenib PI).
NSCLC studies. In patients treated with Combo 450, RPED was reported in 2% (2/98) of patients (one Grade 1 and one Grade 2). Visual impairment, including blurred vision and reduced visual acuity, occurred in 27.6% (27/98) of patients. Visual impairment was generally reversible.
Uveitis was reported when binimetinib was used in combination with encorafenib (see Section 4.8 Adverse Effects (Undesirable Effects) of encorafenib PI).
Left ventricular dysfunction. Melanoma studies. In the pooled Combo 450 population, LVD was reported in 8.4% (23/274) of patients. Grade 3 events occurred in 1.1% (3/274) of patients. LVD led to treatment discontinuation in 0.4% (1/274) of patients and led to dose interruptions or dose reductions in 6.6% (18/274) of patients.
The median time to first occurrence of LVD (any grade) was 4 months (range 0.03 to 21.3 months) in patients who developed an LVEF below 50%. The mean LVEF value dropped by 5.9% in the pooled Combo 450 population from a mean of 63.9% at baseline to 58.1%. LVD was generally reversible following dose reduction or dose interruption.
NSCLC studies. In NSCLC patients treated with Combo 450, LVD was reported in 12.2% (12/98) of patients. Grade 3 events occurred in 2% (2/98) of patients. LVD led to treatment discontinuation in 2% (2/98) of patients and led to dose interruptions or dose reductions in 5.1% (5/98) of patients.
Haemorrhage. Melanoma studies. Haemorrhagic events have been observed in 17.9% (49/274) of patients in the pooled Combo 450 population. Most of these cases were Grade 1 or 2 (14.6%) and 3.3% were Grade 3 or 4 events. Few patients required dose interruptions or dose reductions (0.7% or 2/274). Haemorrhagic events led to discontinuation of treatment in 1.1% (3/274) of patients. The most frequent haemorrhagic events were haematuria in 3.3% (9/274) of patients, rectal haemorrhage in 2.9% (8/274) and haematochezia in 2.9% (8/274) of patients. Fatal gastric ulcer haemorrhage with multiple organ failure as a concurrent cause of death, occurred in one patient. Cerebral haemorrhage occurred in 1.5% (4/274) of patients with fatal outcome in 3 patients. All events occurred in the setting of new or progressive brain metastases for all patients.
In Study CMEK162B2301-Part 2, in the Combo 300 arm, haemorrhagic events were observed in 6.6% (17/257) of patients and were Grade 3-4 in 1.6% (4/257) of patients.
NSCLC studies. Haemorrhagic events have been observed in 13.3% (13/98) of patients with NSCLC treated with Combo 450, including 4.1% of patients with Grade ≥ 3 events. No haemorrhagic events led to discontinuation of treatment in NSCLC population. Dose reduction was required in 5.1% (5/98) of patients. One event of haemorrhage intracranial with fatal outcome (Grade 5) occurred.
Hypertension. Melanoma studies. New onset elevated blood pressure or worsening of pre-existing hypertension were reported in 11.7% (32/274) of patients treated with the Combo 450 mg. Hypertension related adverse events were reported as Grade 3 in 5.5% (15/274) of patients including hypertensive crisis (0.4% (1/274). Hypertension led to dose interruption or adjustment in 2.9% of patients. Hypertensive adverse reactions required additional therapy in 8.0% (22/274) of patients.
NSCLC studies. New onset elevated blood pressure or worsening of pre-existing hypertension were reported in 9.2% (9/98) of patients treated with the Combo 450. Hypertension related adverse reactions were reported as Grade 3 in 4.1% (4/98) of patients. Hypertension did not lead to dose discontinuation, interruption or adjustment.
Venous thromboembolism. Melanoma studies. In the pooled Combo 450 population, VTE occurred in 4.7% (13/274) of patients, including 2.2% (6/274) of patients who developed PE. VTE was reported as Grade 1 or 2 in 3.6% (10/274) of patients and Grade 3 or 4 in 1.1% (3/274) of patients. VTE led to dose interruptions or dose modifications in 1.1% (3/274) patients and to additional therapy in 4.7% (13/274) of patients.
NSCLC studies. In the NSCLC population treated with Combo 450, VTE occurred in 5.1% (5/98) of patients, all events were Grade 2. Including 1.1% (1/98) of patients who developed PE. VTE led to dose interruption or dose modification in 1.0% (1/98) of patient.
Pancreatitis. Pancreatitis was reported when binimetinib was used in combination with encorafenib (see Section 4.8 Adverse Effects (Undesirable Effects) of encorafenib PI).
Dermatological reactions. Rash. Melanoma studies. In the pooled Combo 450 mg population, rash occurred in 19.7% (54/274) of patients. Most of the events were mild, with Grade 3 or 4 events reported in 0.7% (2/274) of patients. Rash led to discontinuation in 0.4% (1/274) patients and to dose interruption or dose modification in 1.1% (3/274) of patients.
NSCLC studies. Rash occurred in 21.4% (21/98) of patients with NSCLC treated with Combo 450 including 2.0% of Grade 3 events.
Palmar-plantar erythrodysaesthesia syndrome. Melanoma studies. Palmar-plantar erythrodysaesthesia syndrome (PPES) was reported when binimetinib was used in combination with encorafenib (see Section 4.8 Adverse Effects (Undesirable Effects) of encorafenib PI).
NSCLC studies. PPES was reported in 2% (2/98) of patients in the Combo 450 NSCLC population (one Grade 1 and one Grade 2). One event led to dose interruption or dose modification, none led to discontinuation.
Dermatitis acneiform. Melanoma studies. In the pooled Combo 450 population, acneiform dermatitis occurred in 4.4% (12/274) of patients with no grade 3/4 events and no event led to treatment discontinuation. Dose modification was reported in 0.7% (2/274) of patients.
NSCLC studies. In the NSCLC population treated with Combo 450, acneiform dermatitis occurred in 3.1% (3/98) of patients including one grade 3 event (1.0%). Acneiform dermatitis did not lead to dose discontinuation, interruption or adjustment.
Photosensitivity. Melanoma studies. In the pooled Combo 450 population, photosensitivity was observed in 4.0% (11/274) of patients. Most events were Grade 1-2, with Grade 3 reported in 0.4% (1/274) of patients and no event led to discontinuation. Dose interruption or dose modification was reported in 0.4% (1/274) of patients.
NSCLC studies. Photosensitivity was reported in 3.1% (3/98) of patients in the Combo 450 NSCLC population (two Grade 1 and one Grade 2). None led to dose interruption, modification or discontinuation.
Facial paresis. Facial paresis was reported when binimetinib was used in combination with encorafenib (see Section 4.8 Adverse Effects (Undesirable Effects) of encorafenib PI).
CK elevation/rhabdomyolysis. Melanoma studies. In the pooled Combo 450 population, mostly mild asymptomatic blood CK elevation was reported in 27.0% (74/274) of patients. The incidence of Grade 3 or 4 adverse events was 5.8% (16/274). The median time to onset of the first event was 2.7 months (range 0.5 to 17.5 months).
Rhabdomyolysis was reported in 0.4% (1/274) of patients treated with encorafenib in combination with binimetinib. In this patient, rhabdomyolysis was observed with concomitant symptomatic Grade 4 CK elevation.
NSCLC studies. In the NSCLC population treated with Combo 450, blood CK elevation was reported in 15.3% (15/98) of patients. The incidence of Grade 3 or 4 adverse events was 3.1% (3/98).
No rhabdomyolysis event was reported in the NSCLC population treated with Combo 450.
Renal dysfunction. Blood creatinine elevation and renal failure occurred when binimetinib was used in combination with encorafenib (see Section 4.8 Adverse Effects (Undesirable Effects) of encorafenib PI).
Liver laboratory abnormalities. Melanoma studies. The incidence of liver laboratory abnormalities reported in the pooled Combo 450 population is listed below:
ALT: 13.1% (36/274) overall - Grade 3-4: 4.7% (13/274);
AST: 9.5% (26/274) overall - Grade 3-4: 2.2% (6/274);
GGT: 14.6% (40/274) overall - Grade 3-4: 8.4% (23/274);
Bilirubin: 0.7% (2/274) overall - the maximum severity of these events was Grade 2.
NSCLC studies. The incidences of liver laboratory abnormalities (increased) reported in the NSCLC population treated with Combo 450 are listed below:
ALT: 13.3% (13/98) overall - Grade 3-4: 5.1% (5/98).
AST: 15.3% (15/98) overall - Grade 3-4: 7.1% (7/98).
GGT: 2.0% (2/98) overall - Grade 3-4: 2.0% (2/98).
Bilirubin 1.0% (1/98) overall - Grade 3-4: 1.0% (1/98).
Gastrointestinal disorders. Melanoma studies. In the pooled Combo 450 population, diarrhoea was observed in 38% (104/274) of patients and was Grade 3 or 4 in 3.3% (9/274) of patients. Diarrhoea led to dose discontinuation in 0.4% of patients and to dose interruption or dose modification in 4.4% of patients. Constipation occurred in 24.1% (66/274) of patients and was Grade 1 or 2. Abdominal pain was reported in 27.4% (75/274) of patients and was Grade 3 in 2.6% (7/274) patients. Nausea occurred in 41.6% (114/274) with Grade 3 or 4 observed in 2.6% (7/274) of patients. Vomiting occurred in 28.1% (77/274) of patients with Grade 3 or 4 reported in 2.2% (6/274) of patients.
Gastrointestinal disorders were typically managed with standard therapy.
NSCLC studies. In the NSCLC population treated with Combo 450, diarrhoea was observed in 52% (51/98) of patients and was Grade 3 in 5.1% of patients (no Grade 4). Diarrhoea led to dose discontinuation in 2.0% of patients and to dose interruption or dose modification in 18.4% of patients. Constipation occurred in 26.5% (26/98) of patients and was Grade 1 or 2. Abdominal pain was reported in 31.6% (31/98) of patients and was Grade 3 in 1.0% of patients. Nausea occurred in 58.2% (57/98) with Grade 3 observed in 4.1% of patients. Vomiting occurred in 39.8% (39/98) of patients and was Grade 3 in 1.0% of patients.
Anaemia. Melanoma studies. In the pooled Combo 450 population, anaemia was reported in 19.7% (54/274) of patients; 4.7% (13/274) of patients had Grade 3 or 4. No patients discontinued treatment due to anaemia, 1.5% (4/274) required dose interruption or dose modification.
In the Combo 300 population of study CMEK162B2301, Part 2, anaemia was observed in 9.7% (25/257) of patients with Grade 3 or 4 reported in 2.7% (7/257) patients.
NSCLC studies. In the NSCLC population treated with the Combo 450, anaemia was reported in 32.7% (32/98) of patients including 13.3% of patients with Grade 3 (no Grade 4). No patients discontinued treatment due to anaemia, 7.1% required dose interruption or dose modification.
Headache. Melanoma studies. In the pooled Combo 450 population, headache occurred in 21.5% (59/274) of patients including Grade 3 in 1.5% (4/274) of patients.
In the Combo 300 population of study CMEK162B2301, Part 2, headache was reported in 12.1% (31/257) of patients and was Grade 3 in 0.4% (1/257) of patients.
NSCLC studies. In the NSCLC population treated with Combo 450, headache was reported in 11.2% (11/98) of patients (only Grade 1 and Grade 2). None led to dose interruption, modification or discontinuation.
Fatigue. Melanoma studies. In the pooled Combo 450 population, fatigue occurred in 43.8% (120/274) of patients including Grade 3 in 2.9% (8/274) of patients.
In the Combo 300 population of study CMEK162B2301, Part 2, fatigue was observed in 33.5% (86/257) of patients with 1.6% (4/257) Grade 3 or 4 events.
NSCLC studies. In the NSCLC population treated with Combo 450, Fatigue was observed in 60.2% (59/98) of patients and was Grade 3 in 8.2% of patients (no Grade 4). Fatigue led to dose discontinuation in 2.0% of patients and to dose interruption or dose modification in 11.2% of patients.
Other adverse reactions. Tumour lysis syndrome (TLS). The occurrence of TLS has been associated with the use of binimetinib in association with encorafenib. The frequency is unknown (see Section 4.4 Special Warnings and Precaution for Use).
Adverse events. Adverse events in melanoma studies. Table 4 summarises adverse events (AEs) occurring at an incidence of ≥ 10% (all grades) or at an incidence of ≥ 2% incidence (grades 3 or 4) and reported in Part 1 of the phase III randomised, active-controlled, open-label, multicentre trial in patients with unresectable or metastatic BRAF V600 E or K mutant melanoma (CMEK162B2301).
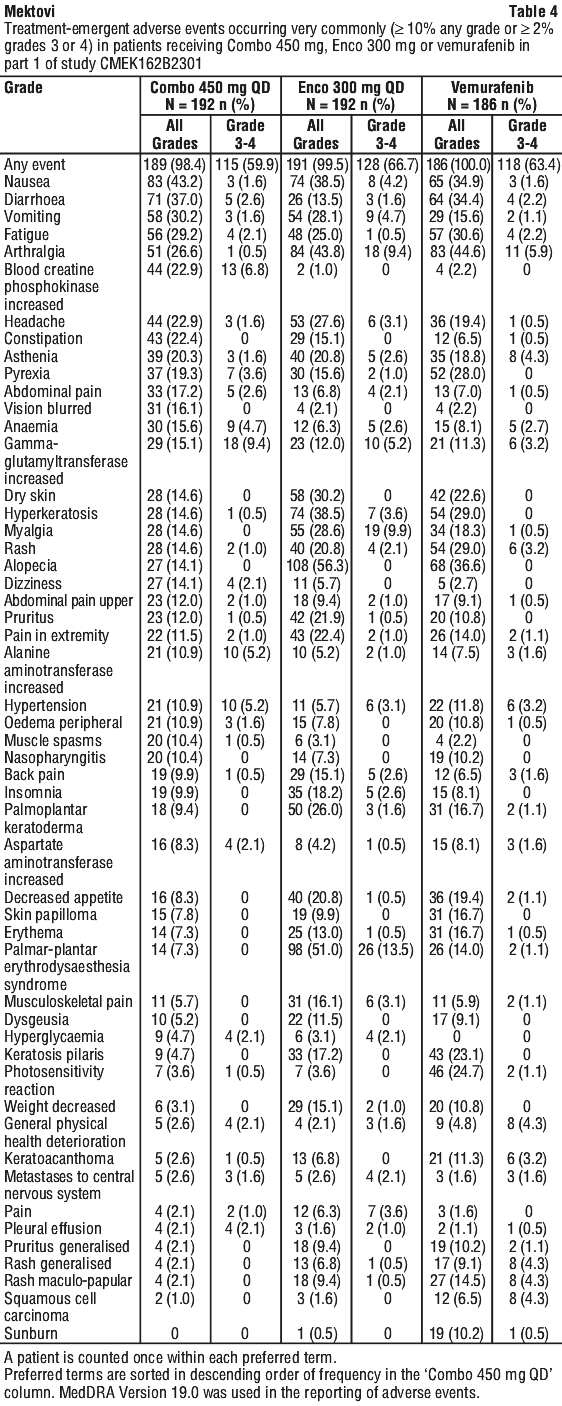
Permanent discontinuations due to AEs were reported in 14.6% of patients treated in the Combo 450 arm, 15.1% of patients treated in the Enco 300 arm and 16.1% of patients treated in the vemurafenib arm.
Adverse events in the NSCLC studies. Table 5 summarises the most common (incidence of at least 10%) treatment-emergent AEs occurring in patients in the PHAROS study. The PHAROS study excluded patients with abnormal left ventricular ejection fraction, prolonged QTc (> 480 ms), uncontrolled hypertension, and history or current evidence of retinal vein occlusion.
Serious adverse reactions occurred in 43.9% of patients who received encorafenib 450 mg in combination with binimetinib. Permanent discontinuations due to AEs were reported in 17.3% of patients treated. The most frequently reported AEs leading to encorafenib discontinuation (≥ 2 (participants) were diarrhoea, nausea, vomiting and myalgia (2.0% each).
Melanoma studies. In patients treated with Combo 450 (n=274), 194 patients (70.8%) were < 65 years, 65 patients (23.7%) were 65-74 years and 15 patients (5.5%) were aged > 75. No overall differences in safety or efficacy were observed between elderly patients (≥ 65) and younger patients. The proportion of patients experiencing adverse events and serious adverse events were similar in patients aged < 65 years and those aged > 65 years. The most common adverse events reported with a higher incidence in patients aged ≥ 65 years compared to patients aged < 65 years included diarrhoea, pruritus, GGT and blood phosphatase alkaline elevation. In the small group of patients aged ≥ 75 years (n=15), patients were more likely to experience serious adverse events and adverse events leading to discontinuation of treatment.
NSCLC studies. In the NSCLC population treated with Combo 450, 36 (36.7%) participants were aged < 65 years and 62 (63.2%) participants were aged ≥ 65 years, including 20.4% aged ≥ 75 years. No notable differences in safety profile were observed between elderly patients (≥ 65) and younger patients.
The most common adverse reactions reported with a higher incidence in patients aged ≥ 75 years compared to patients aged < 75 years included diarrhoea, fatigue and nausea (70.0% each), vomiting (55.0%) and blood creatinine increased (25.0%).
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important.
It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
4.9 Overdose
The highest dose of binimetinib evaluated as single agent in clinical trials was 80 mg administered orally twice daily and was associated with ocular (chorioretinopathy) and skin toxicities (dermatitis acneiform).
In clinical trials of binimetinib in combination with encorafenib, one case of accidental overdose was reported. In this case, a subject took an overdose of 135 mg (9 tablets of binimetinib). No overdose of encorafenib was taken and no adverse events were reported.
Treatment of overdose. There is no specific treatment of overdose. If overdose occurs, the patient should be treated supportively with appropriate monitoring as necessary. Since binimetinib is highly bound to plasma proteins, hemodialysis is likely to be ineffective in the treatment of overdose with binimetinib.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action. Pharmacotherapeutic group: antineoplastic agent, protein kinase inhibitor.
ATC code: L01EE03.
Binimetinib is an ATP-uncompetitive reversible inhibitor of mitogen-activated extracellular signal regulated kinase 1 (MEK1) and MEK2 activation. In a cell free system, binimetinib inhibits MEK1 and MEK2 with the half maximal inhibitory (IC50)'s in the 12-46 nanoM. MEK proteins are upstream regulators of the extracellular signal-related kinase (ERK) pathway, which promotes cellular proliferation. In vitro, binimetinib inhibits MEK-dependent phosphorylation of ERK in human BRAF-mutant melanoma cell lines, significantly inhibiting proliferation and viability of these cell lines. In vivo, binimetinib has been evaluated for its ability to inhibit phosphorylation of ERK and tumour growth in xenograft models in nude mice. Additionally, binimetinib has shown significant anti-tumour activity in BRAF-mutant xenograft models, including melanoma. Overall, binimetinib has demonstrated activity against MEK1 and MEK2 enzymes and possesses anti-proliferative activity in vitro and in vivo.
In non-clinical studies, the combination of binimetinib and encorafenib demonstrated additive or synergistic anti-proliferative activity in vitro in numerous BRAF-mutant cell lines. In vivo, treatment with the combination resulted in greater anti-tumour activity with respect to tumour growth inhibition and better tumour responses (PR and SD) in BRAFV600E mutant human melanoma xenograft studies in mice than that which was achieved with either agent alone.
Clinical trials. COLUMBUS - BRAF V600 mutant unresectable or metastatic melanoma. The safety and efficacy of binimetinib in combination with encorafenib were evaluated in a Phase III, randomised (1:1:1) active-controlled, open-label, multicentre trial in patients with unresectable or metastatic BRAF V600 E or K mutant melanoma (CMEK162B2301). Eligible patients were required to have BRAF V600E or V600K mutation-positive unresectable or metastatic melanoma, as detected using the bioMerieux THxID BRAF assay. Patients were permitted to receive prior adjuvant therapy and one prior line of immunotherapy for unresectable locally advanced or metastatic disease. Prior treatment with BRAF/ MEK inhibitors was not allowed.
Patients included in the study were randomised to receive binimetinib 45 mg orally twice daily plus encorafenib 450 mg orally once daily (Combo 450, N=192), encorafenib 300 mg orally once daily (Enco 300, N=194), or vemurafenib 960 mg orally twice daily (Vem, N=191). Treatment continued until disease progression or unacceptable toxicity.
Randomisation was stratified by American Joint Committee on Cancer (AJCC) Stage (IIIB, IIIC, IVM1a or IVM1b, versus IVM1c), Eastern Cooperative Oncology Group (ECOG) performance status (0 versus 1) and prior immunotherapy for unresectable or metastatic disease (yes versus no).
The primary efficacy outcome measure was progression-free survival (PFS) of Combo 450 compared with vemurafenib as assessed by a blinded independent review committee (BIRC). PFS as assessed by investigators (investigator assessment) was a supportive analysis. The key secondary endpoint included PFS of Combo 450 compared with Enco 300. Other secondary efficacy comparisons between Combo 450 and either vemurafenib or Enco 300 included overall survival (OS), objective response rate (ORR), duration of response (DoR) and disease control rate (DCR) as assessed by BIRC and by investigator assessment.
The median age for patients was 56 years (range 20 - 89), 58% were male, 90% were Caucasian, and 72% of patients had baseline ECOG performance status of 0. Most patients had metastatic disease (95%) and were Stage IVM1c (64%); 27% of patients had elevated baseline serum LDH, and 45% of patients had ≥ 3 organs with tumour involvement at baseline and 3.5% had brain metastases.
A total of 27 patients (5%) had received prior checkpoint inhibitors (anti-PD1/PDL1 or ipilimumab) (8 patients in Combo 450 arm, 4%; 7 patients in vemurafenib arm, 4%; 12 patients in Enco 300 arm, 6%) including 22 patients in the metastatic setting (6 patients in Combo 450 arm; 5 patients in vemurafenib arm; 11 patients in Enco 300 arm) and 5 patients in the adjuvant setting (2 patients in Combo 450 arm; 2 patients in vemurafenib arm; 1 patient in Enco 300 arm).
Most patients were BRAF V600E mutant (88.6%), while the remainder were V600K mutant (10.9%).
The median duration of exposure was 11.7 months in patients treated with Combo 450, 7.1 months in patients with encorafenib 300 mg and 6.2 months in patients with vemurafenib. The median relative dose intensity (RDI) for Combo 450 was 99.6% for binimetinib and 100% for encorafenib the median RDI was 86.2% for Enco 300 and 94.5% for vemurafenib.
Study CMEK162B2301 demonstrated a statistically significant improvement in PFS in patients treated with Combo 450 compared with patients treated with vemurafenib. Patients treated with Combo 450 also had improved ORR, DCR, and DoR compared with patients treated with vemurafenib. Table 6 and Figure 1 summarise the PFS and other efficacy results based on central review of the data by the BIRC.
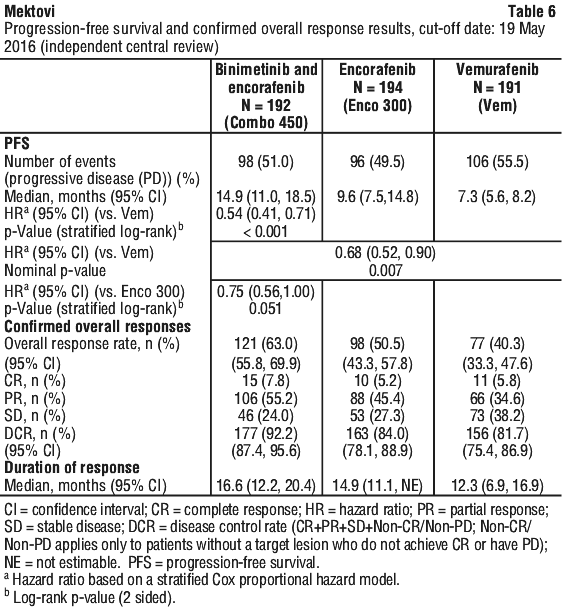
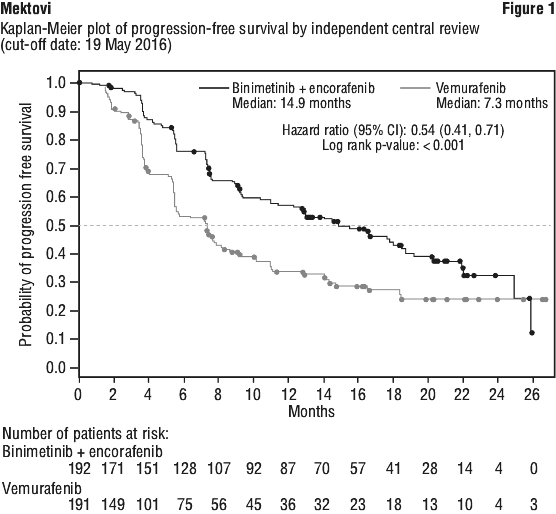
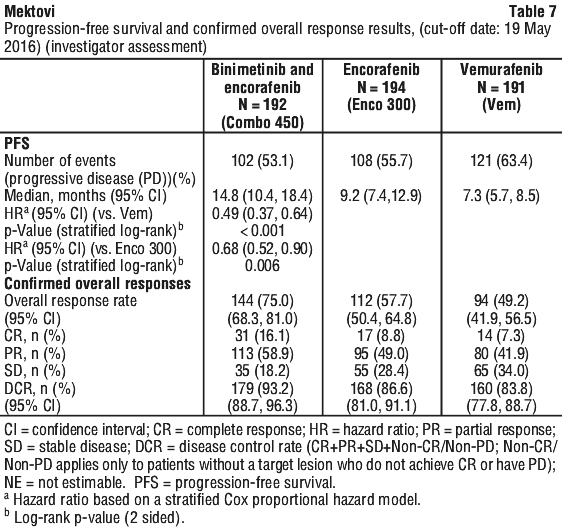
The PFS results per investigator assessment showed consistent results.
An interim OS analysis of study CMEK162B2301 Part 1, performed at the cut-off date of 07 November 2017, demonstrated a statistically significant improvement in OS for Combo 450 compared with vemurafenib (HR 0.61, 95% CI: 0.47, 0.79, see Table 8 and Figure 2). A similar proportion of patients in each treatment arm received subsequent treatment with checkpoint inhibitors, mainly pembrolizumab, nivolumab and ipilimumab (34.4% Combo 450 arm, 36.1% Enco 300 arm, 39.8% vemurafenib arm).
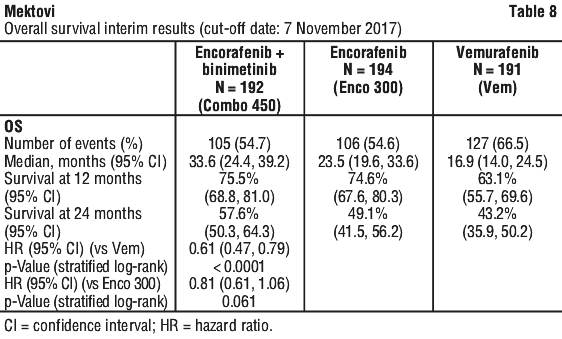
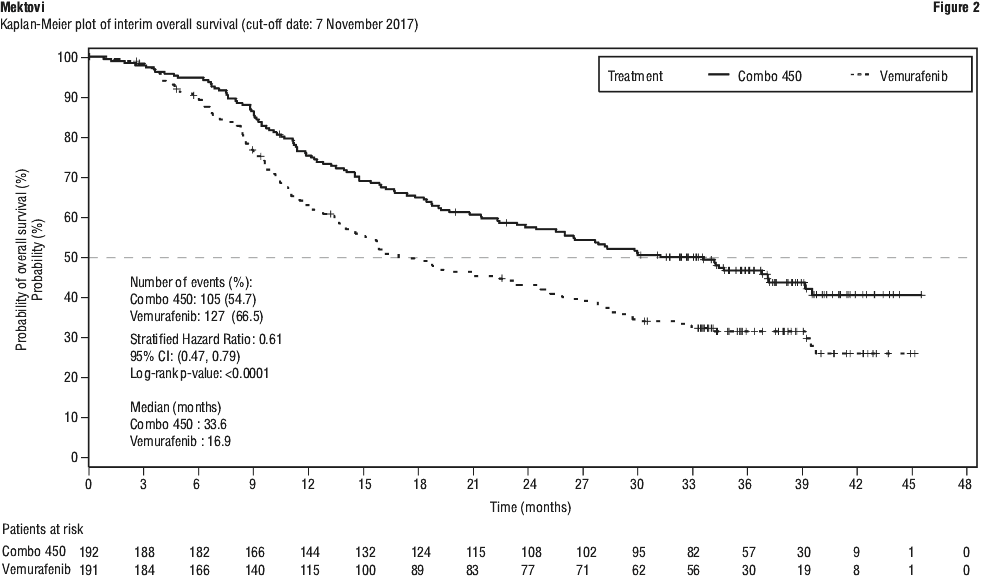
Quality of life (QoL) (cut-off date: 19 May 2016). The Functional Assessment of Cancer Therapy-Melanoma (FACT-M), the European Organization for Research and Treatment of Cancer's core quality of life questionnaire (EORTC QLQ-C30) and the EuroQoL-5 Dimension-5 Level examination (EQ-5D-5L) were used to explore patient-reported outcomes (PRO) measures of health-related Quality of Life, functioning, melanoma symptoms, and treatment-related side effects. The data showed favourable outcomes for the Combo 450 arm over the vemurafenib arm. The median time to definitive 10% deterioration in the FACT-M score was not reached in the Combo 450 arm and was 22.1 months (95% CI: 15.2, NE) in the vemurafenib arm with a HR for the difference of 0.46 (95% CI: 0.29, 0.72). The median time to definitive 10% deterioration in the EORTC QLQ-C30 global health status score was delayed by more than 7 months in the Combo 450 arm compared to the vemurafenib arm: 23.9 months (95% CI: 20.4, NE) vs. 16.6 months (95% CI: 11.9, NE) with a HR for the difference of 0.55 (95% CI: 0.37, 0.80). As these were exploratory endpoints, they must be interpreted with caution in the context of an open-label study design.
Cardiac electrophysiology. In the safety analysis of pooled studies of encorafenib 450 mg once daily in combination with 45 mg binimetinib twice daily, the incidence of new QTcF prolongation > 500 ms was 0.7% (2/268) in the encorafenib 450 mg plus binimetinib group, and 2.5% (5/203) in the encorafenib single agent group. QTcF prolongation of > 60 ms compared to pre-treatment values was observed in 4.9% (13/268) patients in the encorafenib plus binimetinib group, and in 3.4% (7/204) in the encorafenib single agent group (see Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Special Precautions for Use of encorafenib PI).
PHAROS - BRAF V600E mutant advanced non-small cell lung cancer (NSCLC). The safety and efficacy of binimetinib in combination with encorafenib were studied in a Phase II, open-label, multicentre, non-comparative study (Study ARRAY-818-202, PHAROS). Patients were required to have histologically-confirmed metastatic NSCLC with a BRAF V600E mutation, ECOG performance status of 0 or 1, and measurable disease. Patients had received 0 or 1 prior line of systemic therapy in the metastatic setting. Prior use of BRAF inhibitors or MEK inhibitors was prohibited.
Patients were enrolled based on the determination of a BRAF V600E mutation in tumour tissue or blood (e.g. ctDNA genetic testing) by a local laboratory assay. Central confirmation of the BRAF V600E mutation status (i.e. any short variant with protein effect V600E) was performed on archival or fresh tumour tissue collected at enrolment and utilized the FoundationOne CDx - F1CDx (tissue) assay.
A total of 98 patients were enrolled and treated with binimetinib 45 mg orally twice daily and encorafenib 450 mg orally once daily. Treatment continued until disease progression or unacceptable toxicity.
The primary efficacy outcome measure was objective response rate (ORR) and was according to RECIST v1.1 as evaluated by an Independent Radiology Review (IRR). Secondary endpoints included duration of response (DoR), disease control rate (DCR), PFS and OS. Results of the primary analysis with 18.2 months for treatment naïve and 12.8 months previously treated patients are presented below.
Of the 98 patients enrolled in this study, 59 (60.2%) were treatment naïve. The median age of patients was 70 years (47-86), 53% were female, 88% were white and 30% had never smoked. 74% had a baseline ECOG performance status of 1 (67.8 % of participants had a baseline PS 1 in the treatment naïve population and 82.1% in the previously treated population). All patients had metastatic disease of which 8% had brain metastases at baseline and 97% had adenocarcinoma.
At the time of the primary analysis, the median duration of exposure was 15.1 months in treatment naïve patients and 5.4 months in previously treated patients. In the overall population, the median relative dose intensity (RDI) was 95.4% for binimetinib and 99.2% for encorafenib.
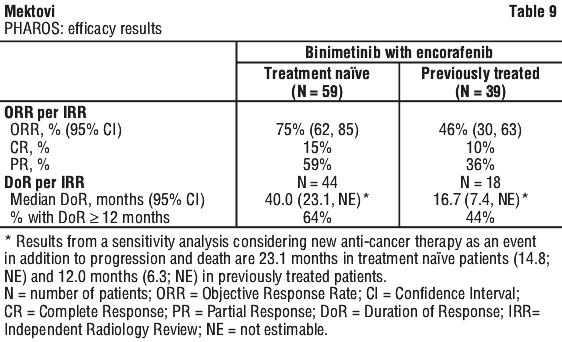
At the time of the primary analysis, the primary endpoint of IRR-assessed ORR in the treatment naïve population was 74.6% (95% CI: 61.6, 85.0), including 9 (15.3%) CRs and 35 (59.3%) PRs. The ORR by IRR in the previously treated population was 46.2% (95% CI: 30.1, 62.8), including 4 (10.3%) CRs and 14 (35.9%) PRs.
Results updated with an additional 10-month follow-up (median duration of exposure of 16.3 months in treatment naïve patients and 5.5 months in previously treated patients) are provided in Table 9.
5.2 Pharmacokinetic Properties
The pharmacokinetics of binimetinib were studied in healthy subjects and patients with solid tumours. After repeated twice-daily dosing concomitantly with encorafenib, steady-state concentrations were reached within 15 days with no major accumulation. The mean (CV %) Cmax,ss was 654 nanogram/mL (34.7%) and mean AUCss was 2.35 microgram.h/mL (28.0%) in combination with encorafenib as estimated by population PK modelling in patients with unresectable or metastatic BRAF V600 mutant melanoma. Binimetinib pharmacokinetics have been shown to be approximately dose-linear.
Absorption. After oral administration, binimetinib is rapidly absorbed with a median Tmax of 1.5 hours. Following a single oral dose of 45 mg [14C] binimetinib in healthy subjects, at least 50% of the binimetinib dose was absorbed. Administration of a single 45 mg dose of binimetinib with a high-fat, high-calorie meal decreased the maximum binimetinib concentration (Cmax) by 17%, while the area under the concentration time curve (AUC) was unchanged. A drug interaction study in healthy subjects indicated that the extent of binimetinib exposure is not altered in the presence of a gastric pH-altering agent (rabeprazole).
Distribution. Binimetinib is 97.2% bound to human plasma proteins in vitro. Binimetinib is distributed to a greater extent in plasma than blood. In humans, the blood-to-plasma ratio is 0.718.
Following a single oral dose of 45 mg [14C] binimetinib in healthy subjects, the apparent volume of distribution (Vz/F) of binimetinib is 374 L.
Metabolism. Following a single oral dose of 45 mg [14C] binimetinib in healthy subjects, the primary biotransformation pathways of binimetinib observed in humans include glucuronidation, N-dealkylation, amide hydrolysis and loss of ethane-diol from the side chain. The maximum contribution of direct glucuronidation to the clearance of binimetinib was estimated to have been 61.2%. Following a single oral dose of 45 mg [14C] binimetinib in healthy subjects, approximately 60% of circulating radioactivity AUC in plasma was attributable to binimetinib. In vitro, CYP1A2 and CYP2C19 catalyses the formation of the active metabolite, which represents < 20% of the binimetinib exposure clinically.
Excretion. Following a single oral dose of 45 mg [14C] binimetinib in healthy subjects, a mean of 62.3% of the radioactivity was eliminated in the faeces while 31.4% was eliminated in urine. In urine, 6.5% of the radioactivity was excreted as binimetinib. The mean (CV %) apparent clearance (CL/F) of binimetinib was 28.2 L/h (17.5%). The median (range) binimetinib terminal half-life (t1/2) was 8.66 h (8.10 to 13.6 h).
Special populations. Hepatic impairment. As binimetinib is primarily metabolised and eliminated via the liver, patients with moderate to severe hepatic impairment may have increased exposure. Results from a dedicated clinical study with binimetinib only indicate similar exposures in patients with mild impairment (Child Pugh Class A) and subjects with normal liver function. A two-fold increase in exposure (AUC) was observed in patients with moderate (Child Pugh Class B) and severe (Child Pugh Class C) hepatic impairment (see Section 4.2 Dose and Method of Administration). This increase expends to three-fold in both moderate and severe hepatic impairment when considering unbound binimetinib exposure (see Section 4.2 Dose and Method of Administration).
The effects of hepatic impairment on the pharmacokinetics of binimetinib in combination with encorafenib have not been evaluated clinically.
Gilbert's syndrome. Binimetinib has not been evaluated in patients with Gilbert's disease. The main route of hepatic transformation of binimetinib being glucuronidation, the decision to treat should be made by the treating physician taking into account the individual benefit-risk.
Renal impairment. Binimetinib undergoes minimal renal elimination. Results from a dedicated clinical trial showed that patients with severe renal impairment (eGFR ≤ 29 mL/min/1.73 m2), had a 29% increase in exposure (AUCinf), a 21% increase in Cmax, and a 22% decrease in CL/F compared to matching healthy subjects. These differences were within the variability observed for these parameters in both cohorts of this study (25% to 49%) and the variability previously observed in patient clinical trials, hence these differences are unlikely to be clinically relevant (see Section 4.2 Dose and Method of Administration).
The effects of renal impairment on the pharmacokinetics of binimetinib in combination with encorafenib have not been evaluated clinically.
Age/body weight. Based on a population PK analysis, age or body weight do not have a clinically important effect on the systemic exposure of binimetinib.
The elderly. Based on results from a population PK analysis of binimetinib in combination with encorafenib, the pharmacokinetics of binimetinib are similar in elderly patients as compared to younger patients.
Paediatric use. The pharmacokinetics of binimetinib have not been established in children and adolescents below the age of 18 years.
Gender. Based on a population PK analysis, the pharmacokinetics of binimetinib were similar in males as compared with females.
Race. There are insufficient data to evaluate potential differences of race or ethnicity on binimetinib pharmacokinetics.
5.3 Preclinical Safety Data
Genotoxicity. Binimetinib was negative for genotoxicity in vitro (reverse mutation in Salmonella typhimurium and E. coli and forward mutation in mouse L5178Y TK+/- lymphoma cells) and in vivo (mouse micronucleus assay).
Carcinogenicity. Carcinogenic potential of binimetinib was not evaluated.
6 Pharmaceutical Particulars
6.1 List of Excipients
The binimetinib drug product is an immediate release film-coated tablet for oral administration. Each tablet contains 15 or 45 mg binimetinib. The tablets also contain the excipients: lactose monohydrate, microcrystalline cellulose, colloidal anhydrous silica, croscarmellose sodium and magnesium stearate in the tablet core; and polyvinyl alcohol, macrogol 3350 (15 mg), macrogol 4000 (45 mg), titanium dioxide (15 mg), purified talc, calcium carbonate (45 mg), iron oxide yellow (15 mg) and iron oxide black (15 mg) in the film coating.
6.2 Incompatibilities
Not applicable.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C.
6.5 Nature and Contents of Container
Mektovi 15 mg film-coated tablets. PVC/PVDC/Aluminium blister containing 12 tablets. Each pack contains 84 tablets.
Mektovi 45 mg film-coated tablets. PVC/PVDC/Alu blister containing 14 tablets.
Each pack contains 28 tablets.
Not all pack sizes may be marketed.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
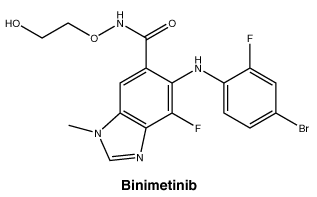
Chemical name. 5-[(4-bromo-2-fluorophenyl)amino]-4-fluoro-N-(2-hydroxyethoxy)- 1-methyl-1H-benzimidazole-6-carboxamide.
Binimetinib is a white to slightly yellow powder with the molecular formula C17H15BrF2N4O3 and a molecular weight of 441.23. At 37°C, binimetinib is slightly soluble (0.1 to 1%) at pH 1.0, very slightly soluble (0.01 to 0.1%) at pH 2.0 and insoluble between pH 4.5 and 7.5. Binimetinib is very slightly soluble (1.6%) in polyethylene glycol, slightly soluble in acetone, acetonitrile, propylene glycol, ethanol and methanol; and very slightly soluble in isopropyl alcohol and n-octanol. Its dissociation constants (pKa) are 2.5 and 8.11. Binimetinib is non-hygroscopic.
7 Medicine Schedule (Poisons Standard)
Schedule 4.
Date of First Approval
03 January 2019
Date of Revision
28 November 2025
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.