Novicrit
Brand Information
| Brand name | Novicrit |
| Active ingredient | Epoetin lambda |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Novicrit.
Summary CMI
Novicrit®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
WARNING: Important safety information is provided in a boxed warning in the full CMI. Read before using this medicine.
1. Why am I using Novicrit?
Novicrit contains the active ingredient epoetin lambda (rch). Novicrit is used to treat anaemia associated with kidney disease, anaemia due to chemotherapy to treat cancer. Novicrit can also be used for patients with mild anaemia who need to donate their own blood before surgery or as alternative to blood transfusion for patients having major bone surgery where high risk of blood transfusion complications.
For more information, see Section 1. Why am I using Novicrit? in the full CMI.
2. What should I know before I use Novicrit?
Do not use if you have ever had an allergic reaction to Novicrit or any of the ingredients listed at the end of the CMI. Use of medicines like Novicrit that stimulate red blood cell production during chemotherapy has been associated with increased risk of death in some studies. Your doctor should only use Novicrit to treat your anaemia if it is caused by chemotherapy and blood transfusions are not an appropriate treatment option. Do not use if you have uncontrolled high blood pressure, been diagnosed with pure red cell aplasia, cannot have transfusions with your own blood, or have severe heart disease, disorders with your veins or arteries, or had a recent heart attack or stroke, or are a surgery patient who should not be given medicines to thin your blood.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use Novicrit? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with Novicrit and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use Novicrit?
Your doctor will determine the correct dose of Novicrit. Novicrit is administered either into a vein (intravenously) or under the skin (subcutaneously).
More instructions can be found in Section 4. How do I use Novicrit? in the full CMI.
5. What should I know while using Novicrit?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using Novicrit? in the full CMI.
6. Are there any side effects?
All medicines can have side effects. Sometimes they are serious, most of the time they are not. If you notice any of the following, tell your doctor immediately as you may need urgent medical care: severe, sudden, stabbing migraine-like headaches. Seizures, confusion or epileptic fits. Raised blood pressure, clotting of your blood in the haemodialysis system, or blockage of your fistula if you are on dialysis. Chest pain, breathlessness, painful swelling in the leg. Signs of an allergic reaction like skin rashes, hives, shortness of breath, wheezing or difficulty breathing, swelling of the face or eyelids. Sudden tiredness, dizziness.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
WARNING FOR CANCER PATIENTS: use of medicines like Novicrit that stimulate red blood cell production during chemotherapy has been associated with increased risk of death in some studies. Your doctor should only use Novicrit to treat your anaemia if it is caused by chemotherapy and blood transfusions are not an appropriate treatment option.
Novicrit®
Active ingredient(s): Epoetin lambda (rch)
Consumer Medicine Information (CMI)
This leaflet provides important information about using Novicrit. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using Novicrit.
Where to find information in this leaflet:
1. Why am I using Novicrit?
2. What should I know before I use Novicrit?
3. What if I am taking other medicines?
4. How do I use Novicrit?
5. What should I know while using Novicrit?
6. Are there any side effects?
7. Product details
1. Why am I using Novicrit?
Novicrit contains the active ingredient epoetin lambda (rch). Novicrit is a protein that stimulates bone marrow to produce more red blood cells. Red blood cells are responsible for carrying oxygen to all parts of your body. A decrease in the number of red blood cells can cause anaemia. Some symptoms of anaemia are tiredness, breathlessness when exercising, and feeling cold. Anaemia may have many causes, including decreased production of a hormone called erythropoietin by the kidneys due to kidney failure, or as a result of chemotherapy treatments for cancer. Novicrit is virtually identical to your body's erythropoietin, and has a similar effect to naturally occurring erythropoietin in your body.
Novicrit is used to treat the anaemia associated with kidney disease. If you have kidney disease, your kidney may not produce enough erythropoietin (necessary for red blood cell production) and your doctor may wish to correct this by prescribing Novicrit. This medicine stimulates your bone marrow to produce more red blood cells, helping to treat your anaemia.
Novicrit can also be used to treat anaemia if you are receiving chemotherapy for cancer and your doctor decides that a blood transfusion is not appropriate.
Doctors can also prescribe Novicrit for mildly anaemic patients who are going to have surgery and donate blood before surgery, so that their own blood can be given to them during or after surgery. Because Novicrit stimulates the production of red blood cells, a higher volume of blood can be taken from these patients.
Novicrit can be used as an alternative to a blood transfusion in adult patients about to undergo major orthopaedic (bone) surgery where there is a potentially high risk from blood transfusion complications.
Novicrit is not addictive.
This medicine is available only with a doctor's prescription.
2. What should I know before I use Novicrit?
Warnings
Do not use Novicrit if:
- you are allergic to epoetin lambda (rch), or any of the ingredients listed at the end of this leaflet.
- Always check the ingredients to make sure you can use this medicine.
- If you have high blood pressure that is not properly controlled with blood pressure-lowering drugs.
- If you are a surgery patient who should not be given medicines to thin the blood.
- If you are due to have elective surgery and you have severe heart disease, disorders of the veins or arteries, or have recently had a heart attack or stroke.
- If you have been diagnosed with Pure Red Cell Aplasia (your bone marrow cannot produce enough red blood cells) after previous treatment with an erythropoietin product, including Novicrit.
- If you cannot have transfusions with your own blood during or after surgery.
- Do not use Novicrit if the packaging is torn or shows signs of tampering.
- Do not use Novicrit beyond the expiry date (month and year) printed on the pack.
- Do not misuse Novicrit. Misuse by healthy people may lead to life-threatening problems with the heart and blood vessels (e.g., stroke, heart attack, or blood clots).
Check with your doctor if you have or have had:
- high blood pressure
- heart disease (such as angina)
- disorders of blood circulation resulting in pins and needles or cold hands or feet or muscle cramps in the legs.
- blood clotting disorders
- seizures or epileptic fits take any medicines for any other condition
- cancer. If you are a cancer patient be aware that erythropoietins like Novicrit may act as a growth factor and therefore in theory may affect the progression of your cancer. Please discuss this with your doctor.
- anaemia from other causes
- liver disease
- gout
- porphyria (a rare blood pigment disorder)
- an allergy to latex.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
In many women with severe kidney failure, their monthly periods may stop. In these women, erythropoietin may restart the monthly cycle. Before starting Novicrit, you should discuss the need for contraception with your doctor.
Make sure you tell your doctor if you have any other medical problems since these may affect the use of Novicrit.
If you have used Novicrit or another erythropoietin in the past, and you lost the good response you were having, tell your doctor about this.
If you have not told your doctor or pharmacist about any of the above, tell them before you start using or are given Novicrit.
Your doctor will advise you whether or not to use Novicrit or if you need to adjust the dose or adapt your treatment.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Iron is also a constituent of red blood cells. Therefore, iron supplements and other blood stimulating drugs may increase your response to Novicrit treatment. Your doctor will decide whether you should take other medicines while using Novicrit.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect Novicrit.
4. How do I use Novicrit?
How much to use
- Your doctor will determine the correct dose of Novicrit. Novicrit is administered either into a vein (intravenously) or just under the skin (subcutaneously). Your doctor can discuss with you whether injection into the vein or under the skin is preferable.
- For patients with anaemia due to kidney failure, Novicrit should be given intravenously, (into a vein or a tube that goes into a vein) if intravenous access is routinely available (haemodialysis patients). For patients not yet on dialysis and those on peritoneal dialysis Novicrit can be administered subcutaneously. The usual starting dose is 50 IU/kg three times per week for adults and 25 IU/kg three times per week for children, after which the dose may be changed by your doctor as needed.
- For patients who are scheduled for surgery and who are not storing their own blood the usual dose is 300 IU/kg body weight for 10 days before surgery, on the day of surgery and for 4 days after. Alternatively a dose of 600 IU/kg may be administered weekly for 3 weeks before surgery and on the day of surgery. The subcutaneous route is used.
- For anaemic cancer patients receiving chemotherapy, the initial dose is 150 IU/kg three times per week. After 4 weeks your doctor will check your response and increase the dose to 300 IU/kg three times weekly if response has been insufficient. If at any stage Novicrit has produced too many red cells, your doctor will stop the drug and later re-start it at a lower dose. The subcutaneous route is used.
When to use Novicrit
Injecting Novicrit under the skin yourself
At the start of your therapy, Novicrit may be injected by medical or nursing staff. However, your doctor may decide that it is right for you to learn how to inject Novicrit under the skin (subcutaneously) yourself. You will receive appropriate training for you to do this.
Under no circumstances should you attempt to inject yourself unless you have been trained to do so.
The Novicrit pre-filled syringe is ready for use. Only use solutions which are clear, colourless and free of visible particles.
Do not shake Novicrit pre-filled syringes.
Ask your doctor if you are unsure of the correct dose for you.
They will tell you exactly how much to use.
Follow the instructions they give you.
If you inject the wrong dose, Epoetin lambda may not work as well and your problem may not improve.
How to inject Novicrit under the skin
For patients with symptomatic anaemia caused by kidney disease, for adult patients receiving chemotherapy, adult patients scheduled for orthopaedic surgery, or adult patients with myelodysplastic syndromes only).
This section contains information on how to give yourself an injection of Novicrit. It is important that you do not try to give yourself the injection unless you have received special training from your doctor or nurse. Novicrit is provided with or without a needle safety guard and you will be shown how to use this by your doctor or nurse. If you are not sure about giving the injection or you have any questions, please ask your doctor or nurse for help.
WARNING: Do not use if the syringe has been dropped onto a hard surface or dropped after removing the needle cap. Do not use the Novicrit prefilled syringe if it is broken. Return the prefilled syringe and the package it came in to the pharmacy.
- Wash your hands.
- Remove one syringe from the pack and remove the protective cap from the injection needle. Syringes are embossed with graduation rings in order to enable partial use if required. Each graduation ring corresponds to a volume of 0.1 mL. If partial use of a syringe is required, remove unwanted solution before injection.
- Clean the skin at the injection site using an alcohol wipe.
- Form a skin fold by pinching the skin between thumb and forefinger.
- Insert the needle into the skin fold with a quick, firm action. Inject the Novicrit solution as you have been shown by your doctor. You should check with your doctor or pharmacist if you are not sure.
Pre-filled syringe without needle safety guard
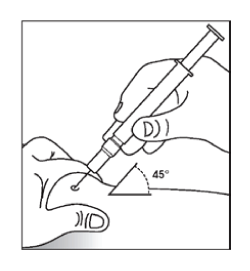
- Always keeping your skin pinched, depress the plunger slowly and evenly.
- After injecting the liquid, remove the needle and let go of your skin. Apply pressure over the injection site with a dry, sterile pad.
- Discard any unused product or waste material. Only use each syringe for one injection.
Pre-filled syringe with needle safety guard
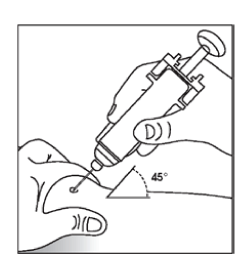
- Always keeping your skin pinched, depress the plunger slowly and evenly until the entire dose has been given and the plunger cannot be depressed any further. Do not release the pressure on the plunger!
- After injecting the liquid, remove the needle while maintaining pressure on the plunger and then let go of your skin. Apply pressure over the injection site with a dry, sterile pad.
- Let go of the plunger. The needle safety guard will rapidly move to cover the needle.
- Discard any unused product or waste material. Only use each syringe for one injection.
If you forget to use Novicrit
Novicrit should be used regularly at the same time.
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Do not take a double dose to make up for the dose you missed.
If you use too much Novicrit
If you think that you have used too much Novicrit, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using Novicrit?
Things you should do
- Always follow your doctor's instructions carefully.
If you are receiving dialysis treatment when you begin treatment with Novicrit, your dialysis regimen may need to be adjusted. Your doctor will decide this.
You will need to have regular blood tests while you are using Novicrit to see how you respond to treatment with Novicrit. Your doctor will order regular blood tests to ensure that your medicine is continuing to work properly. If your haemoglobin levels are above 120 g/L, discuss reducing your Novicrit dose with your doctor.
Your doctor will need to monitor your blood pressure regularly, especially at the beginning of treatment.
An increase in levels of small cells (called platelets) in your blood may occur, particularly when starting haemodialysis treatment.
- Tell your doctor if you become pregnant while using Novicrit.
- If you experience a severe skin reaction, a rash, which may be severe, may cover your whole body and can also include blisters or areas of skin coming off, stop using Novicrit and call your doctor or get medical help right away.
- If you are about to start taking a new medicine, tell your doctor and pharmacist that you are using Novicrit.
- If you become increasingly tired, dizzy or breathless, you should talk to your doctor at once. Your doctor can decide whether Novicrit is not working properly for you and will end the treatment if necessary.
- If you are due to have major surgery, your doctor will give you a medicine to reduce the risk of abnormal blood clotting.
- Remember to tell your doctor if you received Novicrit or another erythropoietin-like medicine in the past and you experienced a worsening in your anaemia.
- Take special care with other products that stimulate red blood cell production: Novicrit is one of a group of products that stimulate the production of red blood cells like the human protein erythropoietin does. If you are given a product in this group other than the one prescribed by your doctor during your treatment, speak to your doctor before using it. It is important that you continue to use the same product in the group unless your doctor says otherwise.
Remind any doctor, dentist or pharmacist you visit that you are using Novicrit.
Things you should not do
- Do not use Novicrit to treat any other complaint unless your doctor says so.
- Do not give this medicine to anyone else, even if their symptoms seem similar to yours.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how Novicrit affects you.
Novicrit may cause dizziness in some people
Looking after your medicine
- Keep the syringes in the original container until it is time to use them.
- Store Novicrit between 2°C and 8°C in the refrigerator. Do not freeze.
- Novicrit may be removed from the refrigerator and stored at up to 25°C for one single period of up to 3 days.
Follow the instructions on the carton on how to take care of your medicine properly.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Keep it where young children cannot reach it.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
Your doctor will want to investigate this. These symptoms may be caused by a condition called pure red cell aplasia (PRCA). PRCA has been rarely reported after months to years of treatment with epoetin. PRCA means the absence of very young red blood cells in the bone marrow. If this condition develops, you suddenly lose the good response you have been having to Novicrit. As the previous cases of PRCA occurred mainly with subcutaneous administration it is preferable that Novicrit be administered intravenously whenever possible. Although PRCA is rare, you should be informed that if it develops, you would need to have regular blood transfusions to treat your anaemia, and Novicrit would have to be stopped. | Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What Novicrit contains
| Active ingredient (main ingredient) | epoetin lambda (rch) |
| Other ingredients (inactive ingredients) | dibasic dihydrate sodium phosphate monobasic dihydrate sodium phosphate sodium chloride glycine polysorbate 80 hydrochloric acid sodium hydroxide water for injection |
| Potential allergens | The needle cover of the pre-filled syringe contains a derivative of latex (FM27) |
Do not take this medicine if you are allergic to any of these ingredients.
What Novicrit looks like
Novicrit 1,000IU/0.5mL - clear, colourless solution, presented in a glass syringe. Each pack contains 1 or 6 syringes (AUST R 147847).
Novicrit 2,000IU/1.0mL - clear, colourless solution, presented in a glass syringe. Each pack contains 1 or 6 syringes (AUST R 147833).
Novicrit 3,000IU/0.3mL - clear, colourless solution, presented in a glass syringe. Each pack contains 1 or 6 syringes (AUST R 147849).
Novicrit 4,000IU/0.4mL - clear, colourless solution, presented in a glass syringe. Each pack contains 1 or 6 syringes (AUST R 147843).
Novicrit 5,000IU/0.5mL - clear, colourless solution, presented in a glass syringe. Each pack contains 1 or 6 syringes (AUST R 147859).
Novicrit 6,000IU/0.6mL - clear, colourless solution, presented in a glass syringe. Each pack contains 1 or 6 syringes (AUST R 147844).
Novicrit 7,000IU/0.7mL - clear, colourless solution, presented in a glass syringe. Each pack contains 1 or 6 syringes (AUST R 147835).
Novicrit 8,000IU/0.8mL - clear, colourless solution, presented in a glass syringe. Each pack contains 1 or 6 syringes (AUST R 147837).
Novicrit 9,000IU/0.9mL - clear, colourless solution, presented in a glass syringe. Each pack contains 1 or 6 syringes (AUST R 147838).
Novicrit 10,000IU/1.0mL - clear, colourless solution, presented in a glass syringe. Each pack contains 1 or 6 syringes (AUST R 147842).
Not all presentations may be available in Australia.
Who distributes Novicrit
Sandoz Pty Ltd
100 Pacific Highway
North Sydney, NSW 2060
Australia
Tel 1800 726 369
This leaflet was prepared in December 2025.
® Registered Trade Mark. The trade marks mentioned in this material are the property of their respective owners.
Brand Information
| Brand name | Novicrit |
| Active ingredient | Epoetin lambda |
| Schedule | S4 |
MIMS Revision Date: 01 December 2024
1 Name of Medicine
Epoetin lambda (rch).
2 Qualitative and Quantitative Composition
The active ingredient of Novicrit Solution for Injection is Epoetin lambda (rch).
Erythropoietin is an endogenous glycoprotein that stimulates red blood cell production. It is normally produced by the kidney and regulated by the level of tissue oxygenation. Epoetin lambda (rch) (CHO) is purified from a Chinese hamster ovary (CHO) cell line into which the gene coding for human erythropoietin has been inserted. Epoetin lambda (rch) is indistinguishable from human erythropoietin in biological activity and immunological reactivity.
Epoetin lambda (rch) has been developed as a similar biological medicinal product to epoetin alfa.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Novicrit is supplied in a pre-filled syringe as a clear, colourless solution for injection.
4 Clinical Particulars
4.1 Therapeutic Indications
Novicrit is indicated for the treatment of patients with symptomatic or transfusion requiring anaemia associated with chronic renal failure to improve their quality of life by improving energy levels, exercise performance, fatigue and sleep patterns and by reducing the need for blood transfusions.
Novicrit is indicated for the treatment of anaemia in patients with nonmyeloid malignancies where anaemia develops as a result of concomitantly administered chemotherapy, and where blood transfusion is not considered appropriate.
Novicrit is also indicated in adult patients with mild to moderate anaemia (haemoglobin > 100 to ≤ 130 g/L) scheduled for elective surgery with an expected moderate blood loss (two to four units or 900 to 1,800 mL) to reduce exposure to allogeneic blood transfusion and to facilitate erythropoietic recovery.
Novicrit is also indicated to augment autologous blood collection and to limit the decline in haemoglobin in anaemic adult patients who are scheduled for major elective surgery and who are not expected to predeposit their complete perioperative blood needs.
4.2 Dose and Method of Administration
Dosage. During therapy, haematological parameters should be monitored regularly. Doses must be individualised to ensure that haemoglobin is maintained at an appropriate level for each patient.
As a single anaphylactic reaction was observed in one patient during the course of clinical testing, it is recommended that the first dose be administered under medical supervision.
Adult patients scheduled for elective surgery. Before considering therapy with Novicrit prior to elective surgery, it is important to investigate and provide appropriate treatment for potentially correctable anaemia.
In patients scheduled for elective surgery, adequate antithrombotic prophylaxis is strongly recommended.
The subcutaneous route of administration should be used.
The recommended dose regimen is 600 IU/kg Novicrit given weekly for three weeks (days -21, -14, and -7) prior to surgery and on the day of surgery. In cases where there is a medical need to shorten the lead time before surgery to less than three weeks, 300 IU/kg Novicrit should be given daily for ten consecutive days prior to surgery, on the day of surgery and for four days immediately thereafter. The administration of Novicrit should be stopped as soon as the haemoglobin level reaches 150 g/L in the preoperative period, even if not all the planned Novicrit doses have been given.
All patients being treated with Novicrit should receive adequate iron supplementation (e.g. oral elemental iron 200 mg daily) throughout the course of Novicrit treatment. If possible, iron supplementation should be started prior to Novicrit therapy, to achieve adequate iron stores.
Anaemic adult surgery patients in an autologous predonation program. The intravenous route should be used. The recommended dose is 300 to 600 IU/kg twice weekly for three weeks, together with at least 200 mg oral elemental iron daily.
Chronic renal failure patients. In patients with chronic renal failure, where intravenous access is routinely available (haemodialysis patients) administration by the intravenous route is preferable. Where intravenous access is not readily available (patient not yet on dialysis and peritoneal dialysis patients) Novicrit may be administered subcutaneously (see Section 4.4 Special Warnings and Precautions for Use, Pure red cell aplasia).
In patients maintained on haemodialysis, Novicrit should always be administered after completion of dialysis.
Treatment with Novicrit is divided into the following two stages:
Correction phase. The initial dose of Novicrit is 50 IU/kg bodyweight three times a week, by intravenous or subcutaneous injection. If the haemoglobin does not increase by 10 g/L after one month of treatment, the dosage may be raised to 75 IU/kg three times/week. If further increments are needed, they should be at 25 IU/kg, three times/week, at monthly intervals, to achieve a haemoglobin not to exceed 120 g/L. This level should not be exceeded in patients with chronic renal failure. Maximum dose should not exceed 3 x 200 IU/kg per week.
Maintenance phase. The intravenous or subcutaneous dose has to be adjusted individually to maintain a haemoglobin not to exceed 120 g/L.
The maintenance dose should be individualised for each chronic renal failure patient. The recommended total weekly dose is between 75 and 300 IU/kg.
For patients who are converted from the subcutaneous route to intravenous route, the same dose should be used, and the haemoglobin should be followed carefully (e.g. weekly) so that appropriate changes in Novicrit dose can be made to keep the haemoglobin within the target range.
Adult patients with cancer. Treatment should not be commenced unless haemoglobin falls below 100 to 110 g/L. The target haemoglobin concentration should be up to 120 g/L in men and women and it should not be exceeded.
The initial dose is 150 IU/kg given subcutaneously three times/week. If the haemoglobin has increased by at least 10 g/L (0.62 mmol/L) or the reticulocyte count has increased ≥ 40,000 cells/microliter above baseline after four weeks of treatment, the dose should remain at 150 IU/kg. If the haemoglobin increase is < 10 g/L (< 0.62 mmol/L) and the reticulocyte count has increased < 40,000 cells/microliter above baseline, increase the dose to 300 IU/kg. If after an additional four weeks of therapy at 300 IU/kg, the haemoglobin has increased ≥ 10 g/L (≥ 0.62 mmol/L) or the reticulocyte count has increased ≥ 40,000 cells/microliter the dose should remain at 300 IU/kg. However, if the haemoglobin has increased < 10 g/L (< 0.62 mmol/L) and the reticulocyte count has increased < 40,000 cells/microliter above baseline, response is unlikely and treatment should be discontinued.
The recommended dosing regimen is described in Figure 1.
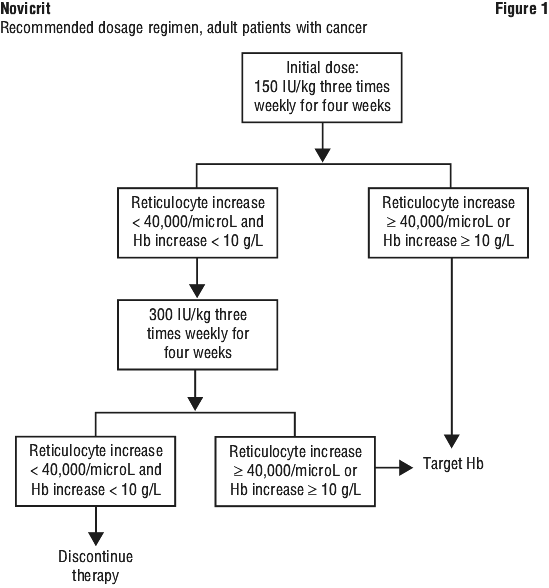
Evaluation of iron status. Iron status should be assessed in all patients prior to therapy. See Section 4.4 Special Warnings and Precautions for Use, Iron supplementation for further information.
Delayed or diminished response. Delayed or diminished response to Novicrit therapy should prompt a search for causative factors such as iron, folate or cyanocobalamin (vitamin B12) deficiency; aluminium intoxication; intercurrent infections; inflammatory or traumatic episodes; occult blood loss; haemolysis; and bone marrow fibrosis of any origin.
Method of administration. Parenteral drug products should be visually inspected for particulate matter and discolouration prior to administration. Product exhibiting particulate matter or discolouration must not be used. Do not shake; shaking may denature the glycoprotein, rendering it inactive.
Syringes are embossed with graduation rings in order to enable partial use if required. Each graduation ring corresponds to a volume of 0.1 mL.
Administer as an intravenous or subcutaneous injection over one to two minutes. In patients on dialysis the injection should follow the dialysis procedure. Slow injection over five minutes may be beneficial to those who experience flu-like symptoms.
Do not dilute or transfer to any other container. Do not administer by intravenous infusion or in conjunction with other medicine solutions.
For the subcutaneous route, a maximum volume of 1 mL at one injection site should generally not be exceeded. In case of larger volumes, more than one site should be chosen for the injection. Subcutaneous injections are given in the limbs or the anterior abdominal wall.
In those situations in which the physician determines that a patient or caregiver can safely and effectively administer Novicrit subcutaneously, instruction as to the proper dose and administration should be provided.
Injecting Novicrit. Do not shake Novicrit syringes. Prolonged vigorous shaking may damage the product. If the product has been shaken vigorously, don't use it.
The Novicrit consumer medicine information includes full instructions for the use and handling of pre-filled syringes.
Dosage adjustment. Renal impairment. If the haemoglobin is increasing and approaching 120 g/L, the dose should be reduced by approximately 25%. If the haemoglobin continues to increase, the dose should be temporarily withheld until the haemoglobin begins to decrease, at which point therapy should be reinitiated at a dose approximately 25% below the previous dose. If the haemoglobin increases by more than 10 g/L in any 2-week period, the dose should be decreased by approximately 25%. If dose reduction is needed the amount given per dose should be reduced or the number of weekly injections reduced or both.
Adult patients with cancer. In oncology patients, rapid increases in haemoglobin concentrations or the use of erythropoietins in subjects with normal haemoglobin concentrations, may result in an increased risk of thrombotic adverse events (see Section 4.4 Special Warnings and Precautions for Use, Cardiovascular and thrombotic events/ increased mortality).
Therefore, a rate of rise in haemoglobin of greater than 10 g/L per two week period or 20 g/L per month, or haemoglobin levels of > 120 g/L should be avoided.
If the haemoglobin is rising by more than 10 g/L per two week period or 20 g/L per month, or haemoglobin is approaching 120 g/L, reduce Novicrit dose by about 25 to 50%. If the haemoglobin exceeds 120 g/L, discontinue therapy until it falls to below 120 g/L and then reinstitute Novicrit at a dose 25% below the previous dose.
4.3 Contraindications
Novicrit is contraindicated in patients with:
Uncontrolled hypertension.
Known sensitivity to mammalian cell derived products.
Hypersensitivity to the active substance or to any of the excipients.
Patients scheduled for elective surgery, who are not participating in an autologous blood predeposit program and who have severe coronary, peripheral arterial, carotid or cerebral vascular disease, including patients with recent myocardial infarction or cerebral vascular accident.
All contraindications associated with autologous blood predonation programmes should be respected in patients being supplemented with epoetin lambda (rch).
Surgery patients who for any reason cannot receive adequate antithrombotic prophylaxis or treatment.
Patients who develop pure red cell aplasia (PRCA) following treatment with any erythropoietin should not receive epoetin lambda (rch) or any other erythropoietin (see Section 4.4 Special Warnings and Precautions for Use).
4.4 Special Warnings and Precautions for Use
Abuse potential. Abuse is the intentional, non-therapeutic use of a drug, even once, for its desirable psychological or physiological effects. Abuse of Novicrit may be seen in athletes for the effects on erythropoiesis. Abuse of drugs that increase erythropoiesis, such as Novicrit, by healthy persons may lead to life-threatening cardiovascular complications (e.g. stroke, myocardial infarction, or thromboembolism).
Cardiovascular and thrombotic events/ increased mortality. An increased incidence of thrombotic vascular events (TVEs) has been observed in patients receiving ESAs such as epoetin lambda (rch) (see Section 4.8 Adverse Effects (Undesirable Effects)). These include venous and arterial thromboses and embolism (including some with fatal outcomes), such as deep venous thrombosis, pulmonary emboli, retinal thrombosis, haemodialysis graft occlusion, myocardial ischaemia and myocardial infarction. Additionally, cerebrovascular accidents (including cerebral infarction, cerebral haemorrhage and transient ischaemic attacks) have been reported (see Section 4.4 Special Warnings and Precautions for Use, Use in renal impairment).
The reported risk of TVEs should be carefully weighed against the benefits to be derived from treatment with epoetin lambda (rch) particularly in patients with pre-existing risk factors for TVE, including obesity and prior history of TVEs (e.g. deep venous thrombosis, pulmonary embolism, and cerebral vascular accident). Epoetin lambda (rch) and other erythropoiesis-stimulating agents increased the risk for death and for serious cardiovascular events in controlled trials when administered to target a haemoglobin of greater than 120 g/L. There was an increased risk of serious arterial and venous thromboembolic events, including myocardial infarction, stroke, congestive heart failure and haemodialysis graft occlusion. A rate of haemoglobin rise of greater than 10 g/L over 2 weeks may also contribute to these risks.
In all patients, haemoglobin levels should be closely monitored due to potential increased risk of thromboembolic events and fatal outcomes when patients are treated at haemoglobin levels above the target range for the indication for use.
Use in cancer patients. In some studies, use of Erythropoiesis Stimulating Agents (ESAs) to treat anaemia in patients with cancer has been associated with increased mortality. ESAs should only be used to treat anaemia that has developed as a result of concomitantly administered chemotherapy, and only when blood transfusion is not considered appropriate. Haemoglobin levels should not exceed 120 g/L.
Epoetin lambda (rch) is a growth factor that primarily stimulates red cell production. Like all growth factors there is a theoretical concern that epoetin lambda (rch) could act as a growth factor for any tumour type, particularly myeloid malignancies.
Cancer patients on Novicrit should have haemoglobin levels measured on a regular basis until a stable level is achieved and periodically thereafter.
As with all growth factors, there is a concern that ESAs could stimulate the growth of tumours. In controlled clinical studies, use of epoetin alfa (rch) and other ESAs have shown:
decreased locoregional control in patients with advanced head and neck cancer receiving radiation therapy when administered to a haemoglobin target of greater than 140 g/L;
shortened overall survival and increased deaths attributed to disease progression at 4 months in patients with metastatic breast cancer receiving chemotherapy when administered to a haemoglobin target of 120-140 g/L.
Another ESA (darbepoetin alfa) increased risk of death when administered to target a haemoglobin of 120 g/L in patients with active malignant disease receiving neither chemotherapy nor radiation therapy. ESAs are not indicated for use in this patient population.
In view of the above, the decision to administer recombinant erythropoietin treatment should be based on a benefit risk assessment with the participation of the individual patient, which should take into account the specific clinical context. Factors to consider in this assessment include: the type of tumour and its stage; the degree of anaemia; life expectancy; the environment in which the patient is being treated; and patient preference.
A study comparing another erythropoiesis stimulating agent with placebo in patients with anaemia of cancer who were not being treated with chemotherapy demonstrated no benefit in terms of reduced transfusion requirements. In addition, there were an increased number of deaths in the active group (26% vs. 20%).
Epoetin lambda (rch) should only be used to treat cancer patients with anaemia where the anaemia has arisen as a result of concomitantly administered chemotherapy. The target haemoglobin should be up to 120 g/L in men and women and it should not be exceeded.
Severe cutaneous adverse reactions. Severe cutaneous adverse reactions (SCARs) including Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), which can be life-threatening or fatal, have been reported in association with epoetin treatment. More severe cases have been observed with long-acting epoetins.
At the time of prescription patients should be advised of the signs and symptoms and monitored closely for skin reactions. If signs and symptoms suggestive of these reactions appear, epoetin alfa should be withdrawn immediately and an alternative treatment considered.
If the patient has developed a severe cutaneous skin reaction such as SJS or TEN due to the use of epoetin alfa, treatment with epoetin alfa must not be restarted in this patient at any time.
Hypertension. Patients with uncontrolled hypertension should not be treated with epoetin lambda (rch); blood pressure should be controlled adequately before initiation of therapy. Blood pressure may rise during treatment of anaemia with epoetin lambda (rch). Hypertensive encephalopathy and seizures have been observed.
Special care should be taken to closely monitor and control blood pressure in patients treated with epoetin lambda (rch). During epoetin lambda (rch) therapy, patients should be advised of the importance of compliance with antihypertensive therapy and dietary restrictions. If blood pressure is difficult to control after initiation of appropriate measures, the dose of epoetin lambda (rch) should be reduced or temporarily withheld until haemoglobin begins to decrease (see Section 4.2 Dose and Method of Administration).
Hypertensive crisis with encephalopathy and seizures, requiring the immediate attention of a physician and intensive medical care, have also occurred during epoetin lambda (rch) treatment in patients with previously normal or low blood pressure. Particular attention should be paid to sudden stabbing migraine-like headaches as a possible warning signal (see Section 4.8 Adverse Effects (Undesirable Effects)).
Pure red cell aplasia. In chronic renal failure patients, antibody mediated pure red cell aplasia (PRCA) (erythroblastopenia) has been rarely reported after months to years of treatment with erythropoietins. Cases also have been rarely reported in patients with hepatitis C treated with interferon and ribavirin, when ESAs are used concomitantly. ESAs are not approved in the management of anaemia associated with hepatitis C.
In most of these PRCA patients, antibodies to erythropoietins have been reported. In patients developing sudden lack of efficacy typical causes of nonresponse should be investigated. If no cause is identified, a bone marrow examination should be considered.
If PRCA is diagnosed, epoetin lambda (rch) must be immediately discontinued and testing for erythropoietin antibodies should be considered. If antibodies to erythropoietin are detected, patients should not be switched to another ESA product as antierythropoietin antibodies cross react with ESAs. Other causes of pure red cell aplasia should be excluded, and appropriate therapy instituted.
PRCA most commonly occurs in patients with chronic renal failure who have received erythropoietins via the subcutaneous route. The subcutaneous route should only be used when intravenous access is not readily available.
No other ESA therapy should be commenced because of the risk of cross reaction.
Seizures. Seizures have occurred in patients with CRF receiving epoetin lambda (rch) with a frequency of 3 to 7%, usually during the first 90 days of treatment. Therefore, Novicrit should be used with caution in patients with epilepsy, history of seizures, or medical conditions associated with a predisposition to seizure activity such as CNS infections and brain metastases. Blood pressure and premonitory neurological symptoms should be closely monitored. Patients should be cautioned to avoid potentially hazardous activities such as driving or operating heavy machinery during this period.
Iron supplementation. Iron status should be assessed in all patients prior to therapy. Further monitoring of serum iron, ferritin and total iron binding capacity is indicated monthly for the first three months of therapy and three monthly thereafter. Virtually all patients will eventually need supplemental iron therapy.
Other causes of anaemia (iron, folate or vitamin B12 deficiency, aluminium intoxication, infection or inflammation, blood loss, haemolysis and bone marrow fibrosis of any origin) should be evaluated and treated prior to initiating therapy with Novicrit, and when deciding to increase the dose. In most cases, the ferritin values in the serum fall simultaneously with the rise in packed cell volume. In order to ensure optimum response to Novicrit, adequate iron stores should be assured and iron supplementation should be administered if necessary.
For chronic renal failure patients, iron supplementation (elemental iron 200-300 mg/day orally for adults and 100-200 mg/day orally for paediatrics) is recommended if serum ferritin levels are below 100 nanogram/mL.
For cancer patients, iron supplementation (elemental iron 200-300 mg/day orally) is recommended if transferring saturation is below 20%.
For patients in an autologous predonation programme, iron supplementation (elemental iron 200 mg/day orally) should be administered several weeks prior to initiating the autologous predeposit in order to achieve high iron stores prior to starting Novicrit therapy, and throughout the course of Novicrit therapy.
For patients scheduled for major elective orthopaedic surgery, iron supplementation (elemental iron 200 mg/day orally) should be administered throughout the course of Novicrit therapy. If possible, iron supplementation should be initiated prior to starting Novicrit therapy to achieve adequate iron stores.
General. Epoetin lambda (rch) should be used with caution in those patients with pre-existing hypertension, ischaemic vascular disease or suspected allergy to any components of the product, porphyria or gout.
The safety and efficacy of epoetin lambda (rch) therapy have not been established in patients with underlying haematological diseases (e.g. haemolytic anaemia, sickle cell anaemia, thalassemia, porphyria).
Erythropoiesis stimulating agents (ESAs) are not necessarily equivalent. Therefore, it should be emphasised that patients should only be switched from one ESA (such as epoetin lambda) to another ESA with the authorisation of the treating physician. In order to improve the traceability of ESAs, the trade name of the administered ESA should be clearly recorded (or stated) in the patient file.
There may be a moderate dose dependent rise in the platelet count within the normal range during treatment with epoetin lambda (rch). This regresses during the course of continued therapy. Development of thrombocytosis is very rare. It is recommended that the platelet count is regularly monitored during the first eight weeks of therapy.
Rarely, development of or exacerbation of porphyria has been observed in epoetin alfa treated patients with chronic renal failure. Epoetin alfa has not caused increased urinary excretion of porphyrin metabolites in normal volunteers, even in the presence of a rapid erythropoietic response. Nevertheless, epoetin lambda (rch) should be used with caution in patients with known porphyria.
Increased serum uric acid may occur in patients whose haemoglobin is rising more than approximately 20 g/L per month. Consequently, epoetin lambda (rch) should be used with caution in patients with a history of gout.
The needle shield on the Novicrit pre-filled syringe contains a derivative of latex (FM27), which may cause allergic reactions in individuals sensitive to latex.
Renal dialysis. Correction of anaemia with epoetin lambda (rch) does not appear to affect dialysis efficiency.
Hyperkalaemia has been observed in isolated cases. However, an increase in appetite could lead to increased potassium intake and hyperkalaemia in both dialysis and predialysis patients. This and other alterations in serum chemistry should be managed by dietary alterations and modifications of the dialysis prescription if appropriate. Serum electrolytes should be monitored in chronic renal failure patients. If an elevated (or rising) serum potassium level is detected then, in addition to appropriate treatment of the hyperkalaemia, consideration should be given to ceasing epoetin lambda (rch) administration until hyperkalaemia has been corrected.
As a result of an increase in packed cell volume, haemodialysis patients receiving Novicrit frequently require an increase in heparin dose during dialysis. If heparinisation is not optimal, occlusion of the dialysis system is possible.
In some female chronic renal failure patients, menses have resumed following epoetin alfa (rch) therapy; the possibility of potential pregnancy should be discussed and the need for contraception evaluated.
In some preclinical toxicological studies in dogs and rats, but not in monkeys, epoetin alfa (rch) therapy was associated with subclinical bone marrow fibrosis. Bone marrow fibrosis is a known complication of chronic renal failure in humans and may be related to secondary hyperparathyroidism or unknown factors. The incidence of bone marrow fibrosis was not increased in a study of dialysis patients who were treated with epoetin alfa (rch) for 12 to 19 months compared to the incidence of bone marrow fibrosis in a matched control group of dialysis patients who had not been treated with epoetin alfa (rch). In a 13 week study, dogs were treated subcutaneously or intravenously with 80, 240 or 520 IU/kg/day. The majority of dogs treated subcutaneously and 50% of dogs treated intravenously developed anaemia with or without bone marrow hypoplasia. The cause of these observations is unknown, however, no cases of paradoxical anaemia have been reported in haematologically normal humans treated with epoetin alfa (rch), making the significance of the findings in dogs unclear.
Use in patients scheduled for elective surgery. Potentially correctable anaemia should be investigated and appropriately treated before considering therapy with epoetin lambda (rch) prior to elective surgery. In patients with a baseline haemoglobin of > 130 g/L (8.1 mmol/L), the possibility that epoetin lambda (rch) treatment may be associated with an increased risk of postoperative thrombotic vascular events cannot be excluded. Therefore, it should not be used in patients with a baseline haemoglobin > 130 g/L (8.1 mmol/L).
Good blood management practices should always be used in the perisurgical setting.
Use in surgery patients in an autologous predonation programme (ABD). All special precautions associated with autologous predonation programs, especially routine volume replacement, should be respected.
Use in hepatic impairment. Novicrit should also be used with caution in patients with chronic liver failure. The safety and dosage regime of epoetin lambda (rch) has not been established in the presence of hepatic dysfunction. Due to decreased metabolism, patients with hepatic dysfunction may have increased erythropoiesis with epoetin lambda (rch).
Use in renal impairment. Chronic renal failure patients being treated with epoetin lambda should have haemoglobin levels measured on a regular basis until a stable level is achieved, and periodically thereafter.
In chronic renal failure patients the rate of increase in haemoglobin should be approximately 10 g/L per month and should not exceed 20 g/L per month to minimise risks of an increase in hypertension. Dose should be reduced when haemoglobin approaches 120 g/L.
In patients with chronic renal failure, maintenance haemoglobin concentration should not exceed the upper limit of the target haemoglobin concentration range as recommended under Dose and Method of Administration. In controlled trials, haemoglobin levels targeted to 130 g/L were associated with a higher risk of cardiovascular or cerebrovascular events, including stroke and death.
Patients with chronic renal failure and insufficient haemogloblin response to ESA therapy may be at even greater risk for cardiovascular or cerebrovascular events and mortality than other patients.
Shunt thromboses have occurred in haemodialysis patients, especially in those who have a tendency to hypotension or whose arteriovenous fistulae exhibit complications (e.g. stenoses, aneurysms, etc). Early shunt revision and thrombosis prophylaxis by administration of acetylsalicylic acid, for example, is recommended in these patients.
Use in the elderly. No data available.
Paediatric use. Efficacy. Clinical trials of epoetin alfa (rch) in children supported the following effects: correction of anaemia; reduction or elimination of transfusion requirements; improvement of the bleeding tendency in uraemia; increased weight and appetite; and the reduction of cytotoxic antibodies. Possible but not conclusive effects were an improvement in exercise capacity and short-term cardiovascular effects. Long-term cardiovascular effects, effects on growth rate, improved prospects for renal transplantation and improved quality of life were unproved.
Safety. Incomplete information is available, particularly on the rate of change of haemoglobin and blood pressure.
Dose. Available data support a dose of 25 IU/kg three times a week rather than 50 IU/kg three times a week.
Effects on laboratory tests. No data available.
4.5 Interactions with Other Medicines and Other Forms of Interactions
There are no known clinically significant interactions with other medicines but the effect of epoetin lambda (rch) may be potentiated by the simultaneous therapeutic administration of a haematinic agent such as ferrous sulfate when a deficiency state exists.
Drugs that decrease erythropoiesis may decrease the response to Novicrit.
Since cyclosporin is bound by red blood cells, there is potential for a medicine interaction. If epoetin lambda (rch) is given concomitantly with cyclosporin, blood levels of cyclosporin should be monitored and the dose of cyclosporin adjusted as the haematocrit rises.
In patients with metastatic breast cancer, subcutaneous co-administration of 40,000 IU/mL epoetin alfa with trastuzumab (6 mg/kg) had no effect on the pharmacokinetics of trastuzumab.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. Intravenous administration of epoetin alfa (rch) at dose levels of 20 to 500 IU/kg/day in rats caused decreased fertility.
Use in pregnancy. (Category B3)
Epoetin lambda (rch) should be administered during pregnancy only if clearly needed and if the potential justifies the potential risk to the foetus. It is not known whether epoetin lambda (rch) crosses the placenta or whether it can cause foetal harm when administered to a pregnant woman. Animal studies have shown no evidence of teratogenic activity in rats or rabbits at epoetin alfa (rch) dosages up to 55 IU/kg/day administered intravenously. However, intravenous administration of epoetin alfa (rch) at dose levels of 20 to 500 IU/kg/day in rats caused decreased fertility, increased preimplantation and postimplantation loss, decreased foetal weight and retardation of ossification.
In pregnant surgical patients participating in an autologous blood predonation program, the use of epoetin lambda (rch) is not recommended.
Use in lactation. Epoetin lambda (rch) should be administered during lactation only if clearly needed. It is not known whether epoetin lambda (rch) is excreted in breast milk or whether it can cause harm to the infant when administered to a lactating woman. Intravenous administration of epoetin alfa (rch) to lactating rats at 500 IU/kg/day causes retardation of growth and development of the offspring.
In lactating surgical patients participating in an autologous blood predonation programme, the use of epoetin lambda (rch) is not recommended.
4.7 Effects on Ability to Drive and Use Machines
Due to the increased risk of hypertension during the initial phase of epoetin lambda (rch) treatment, patients with chronic renal failure should use caution when performing potentially hazardous activities, such as driving or operating machinery, until the optimal maintenance dose of epoetin lambda (rch) has been established.
4.8 Adverse Effects (Undesirable Effects)
Epoetin lambda has been demonstrated to have equivalent pharmacokinetics and pharmacodynamics to epoetin alfa. The adverse effects observed with epoetin alfa would therefore be expected to occur with epoetin lambda.
Adverse effects observed with epoetin alfa. The most frequent adverse drug reaction during treatment with epoetin alfa is a dose dependent increase in blood pressure or aggravation of existing hypertension. Monitoring of the blood pressure should be performed, particularly at the start of therapy. Other common adverse drug reactions observed in clinical trials of epoetin alfa are diarrhoea, nausea, headache, influenza-like illness, pyrexia, rash, and vomiting. Influenza-like illness including headaches, joint pains, myalgia, and pyrexia may occur especially at the start of treatment.
Serious adverse drug reactions include venous and arterial thromboses and embolism (including some with fatal outcomes), such as deep venous thrombosis, pulmonary emboli, arterial thrombosis, retinal thrombosis, and shunt thrombosis (including dialysis equipment). In a cumulative analysis of 10 double blind, randomised, placebo controlled trials in subjects with cancer receiving chemotherapy, deep venous thrombosis was reported in 2.1% and pulmonary embolism in 1.2% of the 1564 subjects exposed to epoetin alfa, compared to 1.2% and 1.2%, respectively, of the 1207 subjects exposed to placebo. Additionally, cerebrovascular accidents (including cerebral infarction and cerebral haemorrhage) and transient ischaemic attacks have been reported in clinical trials of epoetin alfa.
Hypersensitivity reactions, including cases of rash, urticaria, anaphylactic reaction, and angioneurotic oedema have been reported.
Hypertensive crisis with encephalopathy and seizures, requiring the immediate attention of a physician and intensive medical care, have also occurred during epoetin alfa treatment in patients with previously normal or low blood pressure. Particular attention should be paid to sudden stabbing migraine-like headaches as a possible warning signal.
Hyperkalaemia, cough, respiratory congestion, bone pain, porphyria, bone pain in extremity, have also been reported.
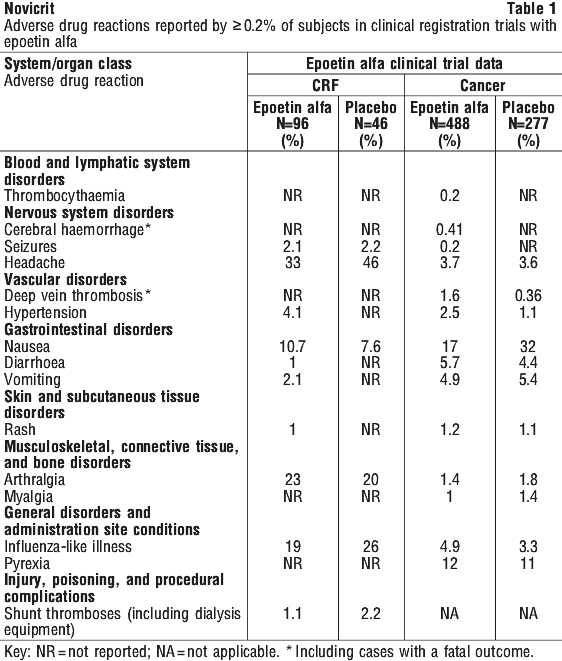
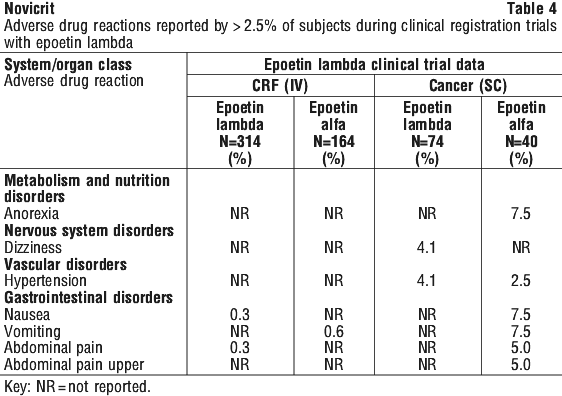
The overall safety profile of epoetin alfa was evaluated in 142 subjects with chronic renal failure (CRF) and in 765 subjects with cancer who participated in placebo controlled, double blind clinical registration trials. Adverse drug reactions reported by ≥ 0.2% of epoetin alfa treated subjects in these trials are shown in Table 1.

Shunt thromboses have occurred in haemodialysis patients, especially in those who have a tendency to hypotension or whose arteriovenous fistulae exhibit complications.
Cancer patients. Thromboembolic events (see Section 4.4 Special Warnings and Precautions for Use) have been reported in cancer patients receiving erythropoietic agents, including epoetin alfa. An investigational study in women with metastatic breast cancer intended to determine whether erythropoietin treatment that extended beyond the correction of anaemia could improve treatment outcomes. However, in that study overall mortality, mortality attributed to disease progression, and incidence of fatal thromboembolic events were all higher in patients receiving epoetin alfa than in those receiving placebo.
Post-marketing data. Adverse drug reactions identified during post-marketing experience with epoetin alfa are included in Table 3. In the table, the frequencies are provided according to the following convention: very common ≥ 1/10; common ≥ 1/100 and < 1/10; uncommon ≥ 1/1,000 and < 1/100; rare ≥ 1/10,000, < 1/1,000; very rare < 1/10,000, including isolated reports.
Antibody mediated pure red cell aplasia has been very rarely reported (< 1/10,000 cases per patient year) after months to years of treatment with epoetin alfa.
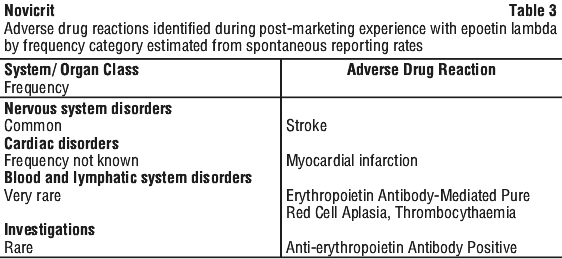
Severe cutaneous adverse reactions (SCARs) including Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), which can be life-threatening or fatal, have been reported in association with epoetin treatment.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
4.9 Overdose
The maximum amount of epoetin lambda (rch) that can be safely administered in single or multiple doses has not been determined with respect to the direct effect of epoetin lambda (rch) as distinct from its effect on red cell mass.
The response to epoetin lambda (rch) is dose related and individual. With excessive erythropoietic response to Novicrit, dosing should be stopped and treatment begun as described above (see Section 4.4 Special Warnings and Precautions for Use, Hypertension, Seizures). Phlebotomy may be performed if excessively high haemoglobin levels occur. Additional supportive care should be provided as necessary.
For information on the management of overdose, contact the Poison Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action. Erythropoietin stimulates erythropoiesis in anaemic patients with chronic renal failure in whom the endogenous production of erythropoietin is impaired. Because of the length of time required for erythropoiesis (several days for erythroid progenitors to mature and be released into the circulation), a clinically significant increase in haemoglobin is usually not observed in less than two weeks and may require up to ten weeks in some patients.
The primary pharmacodynamics of epoetin lambda (rch) were assessed in vitro using an ELISA, by surface plasmon resonance spectroscopy and by use of a cell based assay assessing the response to an erythropoietic stimulus. Comparable responses of epoetin lambda (rch) and the reference product epoetin alfa (rch) were obtained.
The biological efficacy of epoetin lambda (rch) has been demonstrated in vivo using a normocythaemic mouse assay. After administration of epoetin lambda (rch), the reticulocyte counts increased similar to the reference product epoetin alfa.
A 5 day in vivo pharmacodynamic-pharmacokinetic study in Beagle dogs was performed which used reticulocyte pharmacodynamics as biomarker. After three to four days of epoetin lambda (rch) injection a clear rise in reticulocytes was observed, which was reversible upon cessation of treatment. There was no remarkable difference between epoetin lambda (rch) and the reference product epoetin alfa (rch).
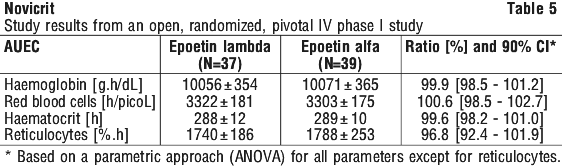
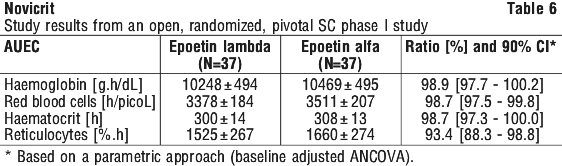
Phase I studies investigating haematological pharmacodynamic parameters following intravenous and subcutaneous single and repeated dosing have demonstrated comparable pharmacodynamics of epoetin lambda (rch) to a reference epoetin alfa (rch) preparation. See Tables 5 and 6.
Epoetin alfa (rch) has been studied in a series of placebo controlled, double blind trials in a total of 131 anaemic cancer patients. Within this group, 72 patients were treated with concomitant noncisplatin containing chemotherapy regimens and 59 patients were treated with concomitant cisplatin containing chemotherapy regimens. Patients were randomised to epoetin alfa (rch) 150 IU/kg or placebo subcutaneously three times a week for 12 weeks.
Epoetin alfa (rch) therapy was associated with a significantly (p < 0.008) greater haematocrit response than in the corresponding placebo treated patients (see Table 7).

Comparable intensity of chemotherapy in the epoetin alfa (rch) and placebo groups in the chemotherapy trials was suggested by a similar area under the neutrophil time curve in patients treated with epoetin alfa (rch) and placebo treated patients as well as by a similar proportion of patients in groups treated with epoetin alfa (rch) and placebo treated groups whose absolute neutrophil counts fell below 1,000 cells/microliter. Available evidence suggests that patients with lymphoid and solid cancers respond equivalently to epoetin alfa (rch) therapy and that patients with or without tumour infiltration of the bone marrow respond equivalently to epoetin alfa (rch) therapy.
Efficacy and safety of epoetin alfa (rch) in the prevention and treatment of anaemia of cancer have not been demonstrated in children.
Epoetin alfa (rch) has been studied in a placebo controlled, double blind trial enrolling 316 patients scheduled for major, elective orthopaedic hip or knee surgery who were expected to require ≥ 2 units of blood. Patients were randomly assigned to receive epoetin alfa (rch) 300 IU/kg, epoetin alfa (rch) 100 IU/kg or placebo by subcutaneous injection for ten days before surgery, on the day of surgery and for four days after surgery. All patients received oral iron and a low dose postoperative warfarin regimen.
Treatment with epoetin alfa (rch) 300 IU/kg significantly (p = 0.024) reduced the risk of allogeneic transfusion in patients with a pretreatment haemoglobin of > 100 to ≤ 130 g/L; 5/31 (16%) of epoetin alfa (rch) 300 IU/kg, 6/26 (23%) of epoetin alfa (rch) 100 IU/kg and 13/29 (45%) of placebo treated patients were transfused.
In the > 100 to ≤ 130 g/L pretreatment stratum, the mean number of units transfused per epoetin alfa (rch) treated patient (0.45 units blood for 300 IU/kg, 0.42 units blood for 100 IU/kg) was less than the mean transfused per placebo treated patient (1.14 units) (overall p = 0.028). In addition, mean haemoglobin, haematocrit and reticulocyte counts increased significantly during the presurgery period in epoetin alfa (rch) treated patients.
Epoetin alfa (rch) was also studied in an open label, parallel group trial enrolling 145 subjects with a pretreatment haemoglobin level of ≥ 100 to ≤ 130 g/L who were scheduled for major orthopaedic hip or knee surgery and who were not participating in an autologous program.
Subjects were randomly assigned to receive one of two subcutaneous dosing regimens of epoetin alfa (rch) (600 IU/kg once weekly for three weeks prior to surgery and on the day of surgery or 300 IU/kg once daily for ten days prior to surgery, on the day of surgery and for four days after surgery). All subjects received oral iron and appropriate pharmacological anticoagulation therapy.
From pretreatment to presurgery, the mean increase in haemoglobin in the 600 IU/kg weekly group (14.4 g/L) was greater than observed in the 300 IU/kg daily group.
The erythropoietic response observed in both treatment groups resulted in similar transfusion rates (11/169 (16%) in the 600 IU/kg weekly group and 14/71 (20%) in the 300 IU/kg daily group). The mean number of units transfused per subject was approximately 0.3 units in both treatment groups.
Using linear logistic models, it can be calculated that for a patient with an entry haemoglobin level of 100 g/L, use of 300 IU/kg daily or 600 IU/kg weekly would reduce the probability of transfusion to about 38%, compared to 58% in the same patient receiving a 100 IU/kg daily regimen, or 81% in a patient given no epoetin alfa (rch) therapy.
Similarly, at a higher entry haemoglobin of 120 g/L, the 300 IU/kg daily or 600 IU/kg weekly regimens would reduce the probability of transfusion to about 18%, compared to 35% in the same patient receiving 100 IU/kg daily, or 61% in a patient receiving no epoetin alfa (rch).
In autologous blood donation, a double blind study was conducted in 204 patients scheduled to undergo elective orthopaedic surgery with haematocrits ≤ 39% and no underlying anaemia due to iron deficiency. On average, patients treated with epoetin alfa (rch) 600 IU/kg twice weekly for three weeks were able to predeposit significantly more units of blood (4.5 units) than placebo treated patients (3.0 units) (p < 0.001). Also, significantly more patients treated with epoetin alfa (rch) (p < 0.05) were able to predeposit between three and six units, inclusively, of autologous blood than the corresponding placebo treated patients. Virtually all (98%) of epoetin alfa (rch) treated patients predeposited three or more units, compared with 69% of placebo treated patients. While 37% of placebo patients were able to predeposit four or five units, 81% of epoetin alfa (rch) patients predeposited four or more units. Among the evaluable patients, fewer patients who received epoetin alfa (rch) required allogeneic transfusions (19.8%) than placebo patients (31%).
In a second placebo controlled study, 55 patients with low haematocrits were enrolled 2:2:1 to receive epoetin alfa (rch) 600 IU/kg, epoetin alfa (rch) 300 IU/kg or placebo twice weekly for three weeks. A significantly greater amount of autologous blood (p < 0.005) was donated by the epoetin alfa (rch) treated patients (4.68 vs. 4.42 vs. 2.89 units). Likewise, 84, 79 and 11% of patients were able to donate four or more units over the three week study.
A randomised, double blind, multicentre phase III study (study number 2003-29-INJ-9) was conducted to evaluate therapeutic equivalence in terms of haemoglobin response of epoetin lambda (rch) versus the reference product epoetin alfa (rch) in the long-term intravenous treatment of anaemia in haemodialysis patients after a 1:1 dose conversion from epoetin alfa (rch) to epoetin lambda (rch).The study included 478 haemodialysis patients with CRF that were treated with the reference product at time of inclusion to the study.
In the first part (double blind) of the study patients were randomly assigned to continue treatment with their original therapy (N = 164) or to switch to epoetin lambda (rch) (N = 314).
In the evaluation phase (weeks 25-28) patients treated with epoetin lambda (rch) showed a comparable Hb level after treatment with epoetin lambda (rch) to their Hb level at start of the treatment. No relevant differences regarding dosing could be observed. Epoetin lambda (rch) has shown to be therapeutically equivalent to epoetin alfa (rch) with respect to Hb response in haemodialysis patients after a 1:1 switch.
In the second (open) part of the study patients of the reference group were changed to epoetin lambda and treated for another 28 weeks. The switch to epoetin lambda did not demonstrate any safety relevant changes.
The long-term safety profiles of epoetin lambda (rch) and epoetin alfa (rch) were comparable. No formation of antiepoetin antibodies was detected. The safety and efficacy of subcutaneous administration of epoetin lambda (rch) in patients with chronic renal failure has not been studied.
A randomised, double blind, multicentre phase III study (study number 2003-31-INJ-11) was conducted to assess the efficacy and safety of epoetin lambda (rch) in the treatment of chemotherapy induced, symptomatic anaemia in patients with solid tumours.
114 patients were treated with epoetin lambda (rch) or epoetin alfa (rch) three times a week subcutaneously for 12 weeks. Doses were raised in case of insufficient increase of Hb respectively reticulocytes after 4 or 8 weeks.
In 62% of patients under epoetin lambda (rch) treatment the Hb level increased by ≥ 20 g/L with the confidence interval being entirely above the predefined threshold of 30%. In the epoetin lambda (rch) group, 32% of patients required transfusions versus 38% in the epoetin alfa (rch) group. None of the secondary efficacy endpoints showed relevant differences between the treatment groups and also the safety profiles were similar.
An open label, single arm, multicentre phase III study (study number HX575-308) was conducted to evaluate the safety and immunogenicity of HX575 epoetin lambda administered subcutaneously in the treatment of anaemia associated with chronic kidney disease in pre-dialysis and dialysis patients.
Patient eligibility was assessed during a 4-week screening period, after which patients entered a 52 week treatment period. A 6-month safety follow-up was conducted for patients with binding, non-neutralising anti-erythropoietin antibodies. Doses were individually titrated to maintain haemoglobin concentrations in the range between 10 and 12 g/dL, and the dosing frequency was adjusted as required. A total of 416 patients were included in the safety population and 303 in the per protocol population.
For ESA naïve patients the starting dose for correcting anaemia was 25 IU/kg of body weight 3 times per week or 75 IU/kg of body weight once per week from analysis week 1 to week 5. After analysis week 5 dose adjustments were possible.
Binding anti-epoetin antibodies were detected via radioimmunoprecipitation (RIP) assay in seven (1.7%, incidence rate of 0.019) of patients in the safety population at some time during the study. No patient developed neutralising antibodies as tested in a cell-based neutralising anti-erythropoietin antibody assay. The 'detected by RIP assay binding antibodies' had no clinical impact on the treatment efficacy. There were no clinical signs of immunogenicity or hypersensitivity.
Prior to study treatment, binding anti-epoetin antibodies were detected in thirteen (eight ESA naïve and 5 ESA pre-treated) of 993 patients (1.3%) via RIP assay during screening. Therefore, given the high sensitivity of the RIP assay, with a potential 1% false-positive rate, the low antibody titres of most positive samples and the fact there was no increase in titer over consecutive visits with continued HX575 treatment, a persistent anti-drug antibody response can be excluded.
Epoetin lambda (rch) was shown to be efficacious in the treatment of chemotherapy associated anaemia in solid tumour patients with a safety profile not differing from what is expected in this therapeutic area.
Epoetin lambda (rch) has not been studied in patients scheduled for elective surgery, either to treat moderate anaemia or to augment autologous blood collection (see Section 4.1 Therapeutic Indications). However, comparable efficacy and safety can be expected in these patients since comparable efficacy and safety to epoetin alfa (rch) has been demonstrated in the anaemia of chronic renal failure (IV administration) and chemotherapy induced anaemia settings.
5.2 Pharmacokinetic Properties
Absorption. Following subcutaneous injection, erythropoietin serum levels remain elevated above baseline for about 72 hours. There is no accumulation when three times weekly dosing is used; the levels remain the same, whether they are determined 24 hours after the first injection or 24 hours after the last injection. Following subcutaneous injection, serum levels are much lower than the levels achieved following intravenous injection; the levels increase slowly and reach a peak between 6 and 24 hours post-dose. The peak is always well below the peak achieved using the intravenous route (approximately 1/20 of the value).
Distribution. The bioavailability of subcutaneous injectable erythropoietin is much lower than that of the intravenous medicine (approximately 20 to 30%).
Metabolism. Metabolism data was not studied for epoetin lambda.
Excretion. After intravenous administration, the mean half-lives in normal volunteers ranged from 2.5 to 6.7 hours. The half-life is difficult to evaluate for the subcutaneous route and is estimated to be about 24 hours.
PK in special populations. No information is available in young or elderly patients. Due to decreased metabolism, patients with hepatic dysfunction may have increased erythropoiesis with epoetin lambda (rch).
Bioequivalence. Phase I studies investigating pharmacokinetic parameters following intravenous and subcutaneous repeated dosing have demonstrated the bioequivalence of Novicrit to a reference epoetin alfa (rch) preparation.
Bioequivalence after multiple intravenous administration was demonstrated in an open, randomised, parallel study in 80 healthy volunteers receiving 100 IU/kg body weight 3 times per week for 4 weeks. The pharmacokinetic parameters are summarised in Table 8.


5.3 Preclinical Safety Data
Genotoxicity. In a standard series of assays for genotoxic potential, epoetin alfa (rch) did not induce gene mutations or cause chromosomal damage.
Carcinogenicity. Long-term carcinogenicity studies have not been carried out. There are conflicting reports in the literature regarding whether erythropoietins may play a role as tumour proliferators. These reports, based on in vitro findings from human tumour samples, are of uncertain significance in the clinical situation.
6 Pharmaceutical Particulars
6.1 List of Excipients
Dibasic dihydrate sodium phosphate, monobasic dihydrate sodium, phosphate, sodium chloride, glycine, polysorbate 80, hydrochloric acid, sodium hydroxide and water for injections. Novicrit pre-filled syringes contain no preservatives.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
For information on interactions with other medicines and other forms of interactions, see Section 4.5.
6.3 Shelf Life
For the purpose of ambulatory use for one single period of up to 3 days.
6.4 Special Precautions for Storage
Store and transport refrigerated (2°C - 8°C). Do not freeze.
Keep the pre-filled syringe in the outer carton in order to protect from light.
For the purpose of ambulatory use, the patient may remove Novicrit from the refrigerator and store it not above 25°C.
6.5 Nature and Contents of Container
Novicrit is supplied as a clear, colourless solution in a single-dose, pre-filled syringe. The prefilled syringes are ready-to-use. The pre-filled syringes are fitted with a needle shield device. The needle cover contains a derivative of latex (FM27) (see Section 4.4 Special Warnings and Precautions for Use).
Packs of 1 syringe or 6 syringes.
Not all presentations are marketed in Australia.
6.6 Special Precautions for Disposal
The pre-filled syringes are for single use in one patient only. Discard any unused portion.
6.7 Physicochemical Properties
Chemical structure. The chemical name of Novicrit is Epoetin lambda (rch). Its empirical formula is C809H1299N229O239S5. (Molecular weight: About 28,000 Daltons. The protein moiety, a single chain polypeptide of 165 amino acids, has a molecular weight of 18,243 Daltons. The carbohydrate moiety with three N-linked and one O-linked carbohydrate groups corresponds to a weight fraction of approximately 40%) and its chemical structure is:
7 Medicine Schedule (Poisons Standard)
S4 - Prescription Only Medicine.
Date of First Approval
27 October 2010
Date of Revision
16 October 2024
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.