Omjjara
Brand Information
| Brand name | Omjjara |
| Active ingredient | Momelotinib |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Omjjara.
Summary CMI
OMJJARA
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
▼ This medicine is new or being used differently. Please report side effects. See the full CMI for further details.
1. Why am I taking OMJJARA?
OMJJARA contains the active ingredient momelotinib, a type of medicine known as a protein kinase inhibitor. OMJJARA is used to to treat adults with some forms of myelofibrosis, a rare form of blood cancer affecting the bone marrow, who also have anaemia (a reduced number of red blood cells), reduce the size of their spleen, and relieve other symptoms related to the disease.
For more information, see Section 1. Why am I taking OMJJARA? in the full CMI.
2. What should I know before I take OMJJARA?
Do not use if you have ever had an allergic reaction to momelotinib or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, had heart trouble or a stroke, have or have had hepatitis B, are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I take OMJJARA? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with OMJJARA and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I take OMJJARA?
- The recommended dosage of OMJJARA for adults is 200 mg taken orally once daily, taken with or without food.
More instructions can be found in Section 4. How do I take OMJJARA? in the full CMI.
5. What should I know while taking OMJJARA?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while taking OMJJARA? in the full CMI.
6. Are there any side effects?
Common side effects include diarrhoea, low platelets (components in the blood that help with clotting), nausea, headache, dizziness, fatigue, feeling weak, abdominal pain, rash and cough.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
▼ This medicine is subject to additional monitoring. This will allow quick identification of new safety information. You can help by reporting any side effects you may get. You can report side effects to your doctor, or directly at www.tga.gov.au/reporting-problems.
OMJJARA
Active ingredient(s): momelotinib
Consumer Medicine Information (CMI)
This leaflet provides important information about using OMJJARA. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using OMJJARA.
Where to find information in this leaflet:
1. Why am I taking OMJJARA?
2. What should I know before I take OMJJARA?
3. What if I am taking other medicines?
4. How do I take OMJJARA?
5. What should I know while taking OMJJARA?
6. Are there any side effects?
7. Product details
1. Why am I taking OMJJARA?
OMJJARA contains the active ingredient momelotinib, which is a type of medicine known as a protein kinase inhibitor.
OMJJARA blocks the action of certain proteins, called Janus Kinases (JAK1, JAK2) and ACVR1. By doing so, it can relieve the symptoms of myelofibrosis. With myelofibrosis, the bone marrow is replaced by scar tissue, and small proteins called cytokines are released in large numbers. The abnormal marrow can no longer produce enough normal blood cells, the spleen can become enlarged, and patients get symptoms such as fever, night sweats, bone pain, and itching.
OMJJARA is used to treat adults with myelofibrosis, a rare form of blood cancer affecting the bone marrow, who also have anaemia (a reduced number of red blood cells), reduce the size of their spleen, and relieve other symptoms related to the disease.
Myelofibrosis may be:
- primary myelofibrosis, which develops in people who have not had problems with their bone marrow before;
- secondary myelofibrosis, which develops in people who have other blood cancers, causing their body to produce too many red blood cells or blood cells called platelets (post polycythaemia vera myelofibrosis or post essential thrombocythaemia myelofibrosis).
2. What should I know before I take OMJJARA?
Things you should do
Before taking OMJJARA, tell your healthcare provider about all of your medical conditions, including if you have had heart trouble or a stroke, have or have had hepatitis B.
Warnings
Do not use OMJJARA if:
- you are allergic to momelotinib, or any of the ingredients listed at the end of this leaflet. Always check the ingredients to make sure you can use this medicine.
Check with your doctor if you:
- have unusual bleeding or bruising under the skin, longer than usual bleeding after your blood has been drawn, or bleeding from your gums - these may be signs of a low blood platelet count.
- have an infection - signs of an infection may include fever, chills, cough, breathing problems, diarrhoea, vomiting, pain or burning feeling when passing urine.
- if you have, or ever had, any liver problems. Your doctor may need to prescribe a different dose of OMJJARA.
- If you had hepatitis B for a long time (chronic) as hepatitis B may become active again.
- have any other medical conditions.
- take any medicines for any other condition.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Blood tests
Before and during treatment, you will have blood tests to check your blood cell levels (red blood cells, white blood cells and platelets) and your liver function. Your doctor may adjust the dose or stop treatment based on the results of the blood tests.
Pregnancy and breastfeeding
If you are pregnant, think you may be pregnant or are planning to have a baby, tell your doctor before you take this medicine as it could harm your baby.
If you are a woman who could become pregnant you must use highly effective contraception while you are taking OMJJARA, and you must continue to use highly effective contraception for at least 1 week after taking your last dose. Your doctor may ask you to take a pregnancy test, to confirm that you are not pregnant, before starting your treatment.
Contact your doctor immediately if you become pregnant while you are taking OMJJARA.
Do not breast-feed while taking OMJJARA. It is not known if it passes into breast milk.
You must stop breast-feeding before taking OMJJARA. Do not begin breast-feeding again until at least 1 week after taking your last dose.
Tell your doctor if you are breast-feeding before taking this medicine.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interfere with OMJJARA and affect how it works.
It is important that you mention any medicines containing any of the following active substances, as your doctor may need to adjust the dose of OMJJARA or the other medicine:
- rosuvastatin (a statin used to lower cholesterol)
- carbamazepine (used to treat epilepsy and control fits or convulsions)
- phenobarbital (used to treat epilepsy and control fits or convulsions)
- phenytoin (used to treat epilepsy and control fits or convulsions)
- St John's wort (Hypericum perforatum), a herbal product
It is currently unknown if OMJJARA could reduce the effectiveness of oral or other contraceptives, therefore it is recommended to add a barrier method during treatment and for at least 1 week after taking your last dose of OMJJARA.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect OMJJARA.
4. How do I take OMJJARA?
How much to take
- The recommended dose of OMJJARA is 200 mg taken by mouth once daily, with or without food.
You will have blood tests before you take this medicine and while you are taking it, to monitor your progress.
If you get certain side effects (such as abnormal bleeding or bruising, diarrhoea or nausea) while you are taking OMJJARA your doctor may recommend a lower dose, or pause or stop your treatment.
Your doctor may recommend a different dose if you have problems with your liver.
When to take OMJJARA
- Take your tablet about the same time once every day.
- Take exactly as your healthcare provider tells you to take it. Check with your doctor or pharmacist if you are not sure.
If you forget to take OMJJARA
OMJJARA should be used regularly at the same time each day. Do not take an extra dose if you miss a dose. Take your next dose at its scheduled time.
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Do not take a double dose to make up for the dose you missed.
If you take too much OMJJARA
If you think that you have used too much OMJJARA, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while taking OMJJARA?
Things you should do
- Always take this medicine exactly as your doctor or pharmacist has told you
Call your doctor straight away if you:
- are pregnant, think you may be pregnant or are planning to have a baby.
- are breastfeeding or intend to breastfeed.
Remind any doctor, dentist or pharmacist you visit that you are using OMJJARA.
Things you should not do
- Do not stop taking OMJJARA unless your doctor tells you to.
- Do not start a new medicine without checking first with the doctor who prescribed OMJJARA.
The following has been observed in another similar type of medicine used for the treatment of rheumatoid arthritis: heart problems, blood clots and cancer.
Talk to your doctor or pharmacist before or during treatment:
- if you are older than 65. Patients aged 65 years and older may be at increased risk of heart problems including heart attack and some types of cancer.
- if you have or have had heart problems.
- if you have or have had cancer.
- if you are a smoker or have smoked in the past.
- if you have previously had blood clots in the veins of your legs (deep vein thrombosis) or lungs (pulmonary embolism) or if you have an increased risk of developing this, for example if:
- you had recent major surgery.
- you use hormonal contraceptives/hormonal replacement therapy.
- you or a close relative have been diagnosed with a blood clotting disorder.
Tell your doctor immediately if you get:
- sudden shortness of breath or difficulty breathing.
- chest pain or pain in upper back.
- swelling of the leg or arm.
- leg pain or tenderness.
- redness or discolouration in the leg or arm.
These can be signs of blood clots in the veins.
- if you notice any new growths on the skin or changes in existing growths. Your doctor may recommend that you have regular skin examinations while taking OMJJARA.
Your doctor will discuss with you if OMJJARA is appropriate for you.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how OMJJARA affects you.
OMJJARA may make you feel dizzy or have blurred vision and therefore influence your ability to drive and use machines. Observe caution when driving or using machines.
Looking after your medicine
- Keep your tablets in the bottle with the desiccant until it is time to take them.
Follow the instructions in the carton on how to take care of your medicine properly.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Keep it where young children cannot reach it.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
Stomach or bowel problems:
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
Infections:
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Other side effects reported (frequency not known)
- allergic reactions (hypersensitivity)
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What OMJJARA contains
| Active ingredient (main ingredient) | momelotinib |
| Other ingredients (inactive ingredients) | microcrystalline cellulose lactose monohydrate sodium starch glycolate (type A) magnesium stearate colloidal anhydrous silica propyl gallate polyvinyl alcohol macrogol 3350 titanium dioxide purified talc iron oxide yellow iron oxide red |
| Potential allergens | lactose monohydrate |
Do not take this medicine if you are allergic to any of these ingredients.
What OMJJARA looks like
OMJJARA 100 mg film-coated tablets
Brown, round tablets, with an underlined “M” debossed on one side and “100” on the other side.
(AUST R 442230)
OMJJARA 150 mg film-coated tablets
Brown, triangle shaped tablets, with an underlined “M” debossed on one side and “150” on the other side.
(AUST R 442231)
OMJJARA 200 mg film-coated tablets
Brown, capsule shaped tablets, with an underlined “M” debossed on one side and “200” on the other side.
(AUST R 442232)
Who distributes OMJJARA
GlaxoSmithKline Australia Pty Ltd
Level 4, 436 Johnston Street,
Abbotsford, Victoria, 3067
Phone: 1800 033 109
www.gsk.com.au
Trade marks are owned by or licensed to the GSK group of companies.
© 2025 GSK group of companies or its licensor.
This leaflet was prepared on 08 December 2025.
Version 2.0
Brand Information
| Brand name | Omjjara |
| Active ingredient | Momelotinib |
| Schedule | S4 |
▼ This medicinal product is subject to additional monitoring in Australia. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse events at www.tga.gov.au/reporting-problems.
MIMS Revision Date: 01 April 2026
1 Name of Medicine
Momelotinib dihydrochloride monohydrate.
2 Qualitative and Quantitative Composition
Each film-coated tablet contains 100 mg, 150 mg or 200 mg of momelotinib as momelotinib dihydrochloride monohydrate.
List of excipients with known effect. Lactose.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Omjjara 100 mg film-coated tablets. Brown, round tablets, with an underlined "M" debossed on one side and "100" on the other side.
Omjjara 150 mg film-coated tablets. Brown, triangle shaped tablets, with an underlined "M" debossed on one side and "150" on the other side.
Omjjara 200 mg film-coated tablets. Brown, capsule shaped tablets, with an underlined "M" debossed on one side and "200" on the other side.
4 Clinical Particulars
4.1 Therapeutic Indications
Omjjara is indicated for the treatment of disease-related splenomegaly or symptoms in adult patients with moderate to severe anaemia who have primary myelofibrosis, post polycythaemia vera myelofibrosis or post essential thrombocythaemia myelofibrosis and who are Janus Kinase (JAK) inhibitor naïve or have been treated with ruxolitinib.
4.2 Dose and Method of Administration
Adults. The recommended dosage of Omjjara is 200 mg taken orally once daily. Omjjara may be taken with or without food.
Missed dose. If a dose of Omjjara is missed, the next scheduled dose should be taken the following day. Two doses should not be taken at the same time to make up for the missed dose.
Monitoring. Complete blood cell count and liver function tests must be performed before initiating treatment with Omjjara, periodically during treatment, and as clinically indicated.
Dose modifications. Dose modifications should be considered for haematologic and nonhaematalogic toxicities (Table 1).
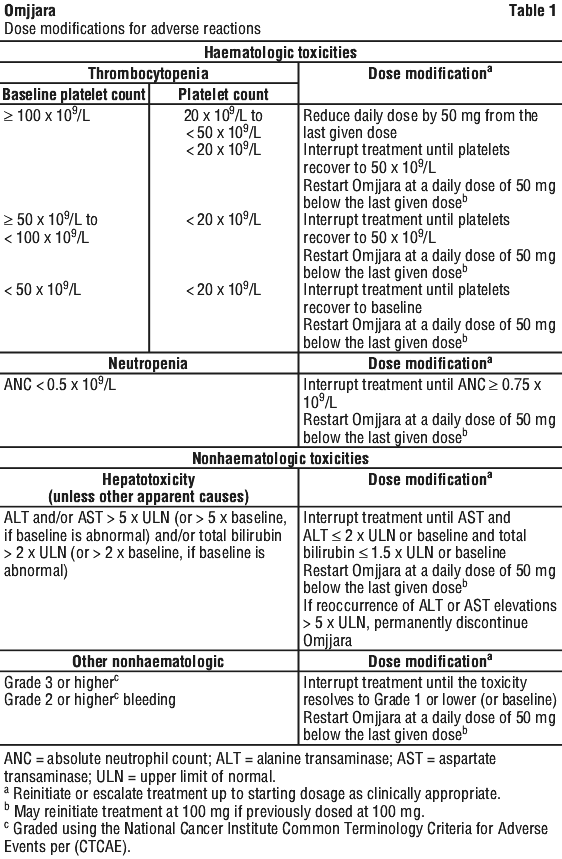
Children. The safety and efficacy of Omjjara in children and adolescents less than 18 years of age have not been established.
Elderly. No dose adjustment is required for patients who are aged 65 years and older (see Section 5.2 Pharmacokinetic Properties, Special patient populations).
Renal impairment. No dose adjustment is required for patients with renal impairment (see Section 5.2 Pharmacokinetic Properties, Special patient populations). Omjjara has not been assessed in patients requiring dialysis.
Hepatic impairment. No dose adjustment is recommended for patients with mild or moderate hepatic impairment. The recommended starting dose of Omjjara is 150 mg once daily in patients with severe hepatic impairment (Child-Pugh Class C) (see Section 5.2 Pharmacokinetic Properties, Special patient populations).
4.3 Contraindications
Omjjara should not be used during pregnancy and breastfeeding.
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of Excipients.
4.4 Special Warnings and Precautions for Use
Infections. Infections, including serious and sometimes fatal bacterial and viral infections (including COVID-19), have occurred in patients treated with Omjjara (see Section 4.8 Adverse Effects (Undesirable Effects)). Omjjara should not be initiated in patients with active infections. Physicians should monitor patients receiving Omjjara for signs and symptoms of infection and initiate appropriate treatment promptly.
Hepatitis B reactivation. Hepatitis B viral load (HBV-DNA titre) increases, with or without associated elevations in alanine transaminase (ALT) or aspartate transaminase (AST), have been reported in patients with chronic hepatitis B virus (HBV) infection taking JAK inhibitors, including Omjjara. The effect of Omjjara on viral replication in patients with chronic HBV infection is unknown. Patients with chronic HBV infection who receive Omjjara should have their chronic HBV infection treated and monitored according to clinical HBV guidelines.
Thrombocytopenia and neutropenia. New onset of severe (Grade ≥ 3) thrombocytopenia and neutropenia was observed in patients treated with Omjjara (see Section 4.8 Adverse Effects (Undesirable Effects)). A complete blood count, including platelet count, should be obtained before initiating treatment with Omjjara, periodically during treatment, and as clinically indicated. Dose interruption or reduction may be required (see Section 4.2 Dose and Method of Administration, Dose modifications).
Major adverse cardiovascular events (MACE). In a large randomised active-controlled study of tofacitinib (another JAK inhibitor) in rheumatoid arthritis patients 50 years and older with at least one additional cardiovascular risk factor, a higher rate of MACE, defined as cardiovascular death, non-fatal myocardial infarction (MI) and non-fatal stroke, was observed with tofacitinib compared to tumour necrosis factor (TNF) inhibitors.
Events of MACE have been reported in patients receiving Omjjara, however, a causal relationship has not been established. Prior to initiating or continuing therapy with Omjjara, the benefits and risks for the individual patient should be considered particularly in patients 65 years of age and older, patients who are current or past long-time smokers, and patients with history of atherosclerotic cardiovascular disease or other cardiovascular risk factors.
Thrombosis. In a large randomised active-controlled study of tofacitinib (another JAK inhibitor) in rheumatoid arthritis patients 50 years and older with at least one additional cardiovascular risk factor, a dose dependent higher rate of venous thromboembolic events (VTE) including deep venous thrombosis (DVT) and pulmonary embolism (PE) was observed with tofacitinib compared to TNF inhibitors.
Events of DVT and PE have been reported in patients receiving Omjjara. However, a causal association has not been established. In patients with myelofibrosis treated with Omjjara in clinical trials, the rates of thromboembolic events were similar in Omjjara and control-treated patients. Prior to initiating or continuing therapy with Omjjara, the benefits and risks for the individual patient should be considered particularly in patients with cardiovascular risk factors (see Section 4.4 Special Warnings and Precautions for Use, Major adverse cardiovascular events (MACE)).
Patients with symptoms of thrombosis should be promptly evaluated and treated appropriately.
Second primary malignancies. In a large randomised active controlled study of tofacitinib (another JAK inhibitor) in rheumatoid arthritis patients 50 years and older with at least one additional cardiovascular risk factor, a higher rate of malignancies, particularly lung cancer, lymphoma and non-melanoma skin cancer (NMSC) was observed with tofacitinib compared to TNF inhibitors.
Lymphoma and other malignancies have been reported in patients receiving JAK inhibitors, including Omjjara. However, a causal association has not been established.
Women of childbearing potential. Given uncertainties about whether Omjjara may reduce the effectiveness of hormonal contraceptives, women using oral or other hormonal contraceptives should add a barrier method during treatment and for at least 1 week after the last dose of Omjjara.
Use in hepatic impairment. See Section 5.2 Pharmacokinetic Properties, Special patient populations.
Use in renal impairment. See Section 5.2 Pharmacokinetic Properties, Special patient populations.
Use in the elderly. See Section 5.2 Pharmacokinetic Properties, Special patient populations.
Paediatric use. The safety and efficacy of Omjjara in children and adolescents less than 18 years of age has not been established.
Effects on laboratory tests. No data available.
4.5 Interactions with Other Medicines and Other Forms of Interactions
Effect of other medicinal products on momelotinib. Effect of CYP inhibitors/ inducers on momelotinib. Momelotinib undergoes metabolism through multiple CYP enzymes (including CYP3A4, CYP2C8, CYP2C9, CYP2C19, and CYP1A2), with CYP3A4 having the greatest contribution.
Strong CYP3A4 inducers. Coadministration of strong CYP3A4 inducers may lead to decreased exposure of Omjjara and consequently a risk for reduced efficacy. Therefore, additional monitoring is recommended with concomitant use of Omjjara and strong CYP3A4 inducers (including but not limited to carbamazepine, phenobarbital, phenytoin, and St John's wort [Hypericum perforatum] (see Section 5.2 Pharmacokinetic Properties).
Effect of transporters inhibitors/ inducers on momelotinib. Momelotinib and its active metabolite, M21, are substrates of P-glycoprotein, BCRP, OATP1B1 and OATP1B3 transporters. Inhibitors or inducers of these transporters may potentially alter the systemic exposure of momelotinib and M21.
Effect of momelotinib on other medicinal products. Effect of momelotinib on CYP enzymes. In vitro studies indicate that momelotinib may inhibit CYP2B6 while M21 does not. Additionally, both momelotinib and M21 do not inhibit CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6 or CYP3A4 in vitro. Furthermore, momelotinib and M21 do not induce CYP3A4 in vitro.
Effect of momelotinib on UGT enzymes. In vitro, momelotinib, but not M21, is an inhibitor of UGT1A1 and UGT1A9. Momelotinib and M21 are not inhibitors of UGT1A3, UGT1A4, UGT1A6 or UGT2B7.
Effect of momelotinib on drug transporters. Momelotinib is an inhibitor of BCRP, and M21 is an inhibitor of MATE1 in vitro. Momelotinib may increase the plasma concentration of medicinal products that are sensitive BCRP and MATE1 substrates; patients should be monitored for adverse reactions with co-administration.
In vitro, neither momelotinib nor M21 inhibits OAT1, OAT3 and OCT2 at clinically-relevant concentrations. Momelotinib and M21 have not been evaluated for MATE2-K inhibition.
Effect of momelotinib on hormonal contraceptives. It is currently unknown whether momelotinib may reduce the effectiveness of systemically acting hormonal contraceptives. Therefore, women using systemically acting hormonal contraceptives should add a barrier method during Omjjara treatment and for at least 1 week after the last dose (see Section 4.6 Fertility, Pregnancy and Lactation).
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. There are no data on the effects of Omjjara on human male or female fertility.
In male rats, momelotinib reduced sperm concentration and motility and reduced testes and seminal vesicle weights at oral doses ≥ 25 mg/kg/day (yielding 16-fold the plasma AUC for combined momelotinib and M21 [minimally produced in rats] in patients at the maximum recommended human dose [MRHD] of 200 mg), leading to reduced fertility at 68 mg/kg/day (44-fold he plasma AUC for combined momelotinib and M21 in patients at the MRHD).
Momelotinib impaired female fertility at oral doses of 68 mg/kg/day (yielding 59-fold the plasma AUC for combined momelotinib and M21 [minimally produced in rats] in patients at the MRHD), with reduction in the number of corpora lutea, oestrous cycles and implantation sites. Oral doses of ≥ 25 mg/kg/day (yielding 22-fold the plasma AUC for combined momelotinib and M21 in patients at the MRHD) caused adverse effects on early embryonic development (pre- and post-implantation loss, reduced number of live fetuses and total litter loss) in rats. Exposures at the no-observed-adverse-effect-level (NOAEL) in male and female rats at 5 mg/kg/day were approximately 3 times the recommended dose of 200 mg daily (based on combined momelotinib and M21 AUC).
Use in pregnancy. (Category D)
There are no data on the effects of Omjjara in human pregnancy to inform a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes.
Embryofetal toxicity (embryonic death, visceral malformation, skeletal variations, and lower mean fetal body weights) was observed at oral maternal doses of 12 mg/kg (yielding exposure based on combined momelotinib and M21 [a major active human metabolite, minimally produced in rats] AUC 10-fold higher than in patients at the MRHD). Skeletal variations were observed (in the absence of maternal toxicity) at 6 mg/kg/day (resulting in exposures 3.5-fold higher than in patients at the MRHD based on combined momelotinib and M21 AUC). Abortion, reduced fetal weight and increased incidence of delayed ossification were observed in rabbits at 60 mg/kg/day (at exposures less than in patients at the MRHD based on combined momelotinib and M21 [minimally produced in rabbits] AUC), occurring in the context of maternotoxicity. No adverse effects on embryofetal development were observed in rats at 2 mg/kg/day and in rabbits at 30 mg/kg/day (exposures less than the exposure at the MRHD). In a pre- and postnatal development study in rats, momelotinib oral administration from organogenesis through lactation reduced the number of live pups at 12 mg/kg/day and lowered postnatal survival and offspring body weight at ≥ 6 mg/kg/day (at exposures less than in patients at the MRHD based on combined momelotinib and M21 AUC).
Omjjara should not be used during pregnancy or by women attempting to become pregnant. Females of reproductive potential who are not pregnant should use highly effective contraception during therapy and for at least 1 week after the last dose of Omjjara.
Use in lactation. There are no data on the presence of momelotinib and/or its metabolites in human milk. It is not known if momelotinib and/or its metabolites are excreted in human milk. Momelotinib was present in rat pups following nursing from treated dams (likely due to the presence of momelotinib in milk) with adverse effects in the offspring. A risk to the breastfed child cannot be excluded.
Patients should not breastfeed during treatment with Omjjara and for at least 1 week after the last dose of Omjjara.
4.7 Effects on Ability to Drive and Use Machines
There have been no studies to investigate the effect of Omjjara on driving performance or the ability to operate machinery. However, patients who experience dizziness or blurred vision after taking Omjjara should observe caution when driving or using machines (see Section 4.8 Adverse Effects (Undesirable Effects)).
4.8 Adverse Effects (Undesirable Effects)
The safety of Omjjara, based on three randomised, active-controlled, multicentre studies in adults with myelofibrosis (MOMENTUM, SIMPLIFY-1, SIMPLIFY-2), is presented in Table 2. Subjects were initially randomised to receive Omjjara 200 mg once daily for 24 weeks (n = 448). The adverse reactions identified for Omjjara are listed by body system organ class (SOC) and frequency.
Frequencies are defined as: very common: ≥ 1/10; common: ≥ 1/100 to < 1/10; uncommon: ≥ 1/1,000 to < 1/100; rare: ≥ 1/10,000 to < 1/1,000.
The most common severe adverse reaction (≥ Grade 3) was thrombocytopenia (12%). The most common adverse reaction leading to discontinuation of Omjjara was thrombocytopenia (2.5%). The most common adverse reaction requiring dosage reduction and/or treatment interruption was thrombocytopenia (7%).
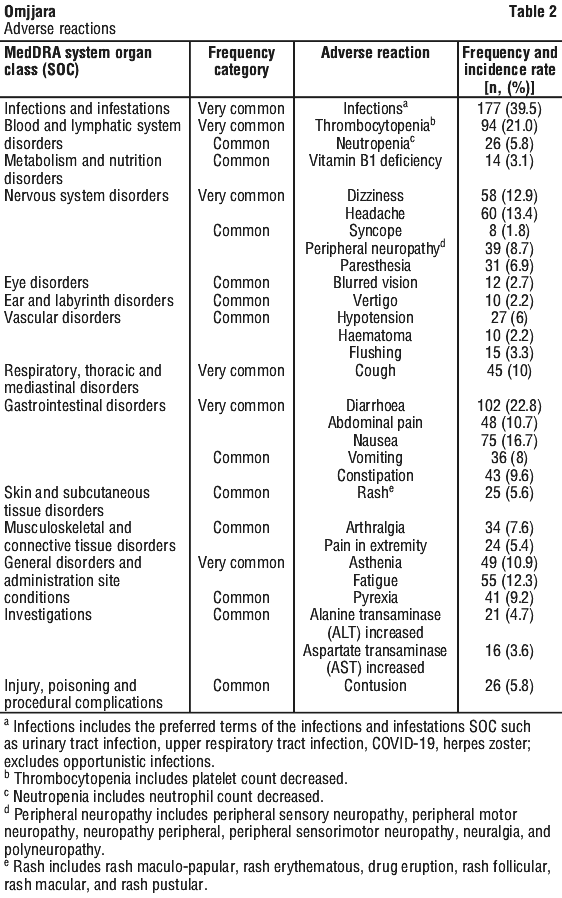
Thrombocytopenia. In the three randomised clinical studies, 21% (94/448) of patients treated with Omjjara experienced thrombocytopenia; 12% (54/448) of patients treated with Omjjara experienced severe thrombocytopenia (≥ Grade 3). The proportion of patients discontinuing treatment due to thrombocytopenia was 2.5% (11/448). In the individual studies of MOMENTUM, SIMPLIFY-1, and SIMPLIFY-2, the rates of thrombocytopenia for Omjjara were 28%, 19%, and 17%, respectively, compared with 15% for danazol, 29% for ruxolitinib, and 12% for best available therapy.
Elevated ALT/AST. In the three randomised clinical trials, new or worsening elevations of ALT and AST (all grades) occurred in 20% (88/448) and 20% (90/448), respectively, of patients treated with Omjjara; Grade 3 and 4 transaminase elevations occurred in 1.1% (5/448) and 0.2% (1/448) of patients, respectively. Reversible drug-induced liver injury has been reported in patients with myelofibrosis treated with Omjjara in clinical trials.
Post-marketing data. As these post-marketing events were reported from a population of unknown size, the frequency is unknown.
Immune system disorders. Hypersensitivity.
Skin and subcutaneous tissue disorders. Rash: cases of rash requiring hospitalisation have been reported.
Erythema multiforme.
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
4.9 Overdose
There is currently limited experience of overdosage with Omjjara. If overdose is suspected, the patient should be monitored for any signs or symptoms of adverse reactions or effects, and appropriate standard of care measures should be instituted immediately. Further management should be as clinically indicated or as recommended by the national poisons centre, where available.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action. Myelofibrosis is a myeloproliferative neoplasm associated with constitutive activation and dysregulated Janus Kinase (JAK) signalling that contributes to elevated inflammation and hyperactivation of activin A receptor type 1 (ACVR1), also known as activin receptor-like kinase 2 (ALK2). Momelotinib and its major circulating metabolite (M21) are inhibitors of wild type Janus Kinase 1 and 2 (JAK1/JAK2) and mutant JAK2V617F, which contribute to signalling of a number of cytokines and growth factors that are important for haematopoiesis and immune function. JAK1 and JAK2 recruit and activate STAT (signal transducer and activator of transcription) proteins that control gene transcription impacting inflammation, haematopoiesis, and immune regulation. Momelotinib and its major human circulating metabolite, M21, additionally inhibit ACVR1 which subsequently down regulates liver hepcidin expression resulting in increased iron availability and red blood cell production. Momelotinib and M21 potentially inhibit additional kinases, such as other JAK family members, IκB kinase (IKK), interleukin-1 receptor-associated kinase 1 (IRAK1), and others.
Pharmacodynamic effects. Omjjara inhibits cytokine-induced STAT3 phosphorylation in whole blood from patients with myelofibrosis. Maximal inhibition of STAT3 phosphorylation occurred 2 hours after Omjjara dosing with inhibition persisting for at least 6 hours. Omjjara also demonstrated both acute and prolonged reduction of circulating hepcidin in patients with myelofibrosis, resulting in increased iron availability and erythropoiesis.
Cardiovascular effects. At a dose of 4 times the highest recommended starting dosage of 200 mg, Omjjara did not prolong the QT interval to any clinically relevant extent.
Clinical trials. The efficacy of Omjjara in the treatment of patients with intermediate-1, intermediate-2, or high-risk myelofibrosis, including primary myelofibrosis, post-polycythaemia vera (post-PV) myelofibrosis or post-essential thrombocythaemia (post-ET) myelofibrosis, was established in two randomised, active-controlled Phase 3 studies, MOMENTUM and SIMPLIFY-1. All patients received a starting dose of Omjjara 200 mg once daily, irrespective of their baseline platelet count (in MOMENTUM study, the minimum platelet count was 25 x 109/L; in SIMPLIFY 1 study, the minimum platelet count was 50 x 109/L).
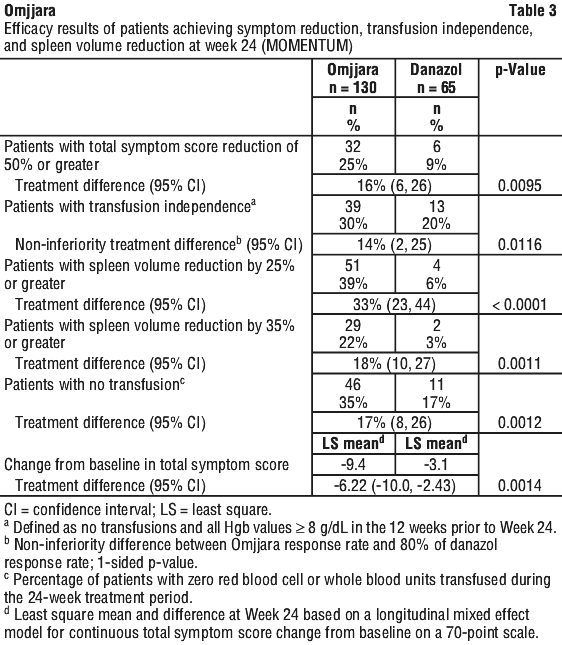
MOMENTUM. Myelofibrosis patients who have been treated with ruxolitinib. MOMENTUM was a double-blind, 2:1 randomised, active-controlled study in 195 symptomatic and anaemic patients with myelofibrosis who had previously received a JAK inhibitor. The median age was 71 years (range 38 to 86 years); 79% were 65 years or older and 63% were male. Sixty-four percent (64%) of patients had primary myelofibrosis, 19% had post-PV myelofibrosis, and 17% had post-ET myelofibrosis. Five percent (5%) of patients had intermediate-1 risk, 57% had intermediate-2 risk, and 35% had high-risk disease. Patients were symptomatic with a Myelofibrosis Symptom Assessment Form (MFSAF) v4.0 total symptom score (TSS) of ≥ 10 at screening (mean MFSAF TSS 27 at baseline), and anaemic with haemoglobin (Hgb) < 10 g/dL. The MFSAF daily diary captured the core symptoms of myelofibrosis: night sweats, abdominal discomfort, pain under the left rib, fatigue/tiredness, early satiety, pruritus, and bone pain. Within the 8 weeks prior to enrolment, 79% had red blood cell transfusions. At baseline, 13% and 15% of patients were transfusion independent in the Omjjara and danazol groups, respectively. The baseline median Hgb was 8 g/dL and the median platelet count was 96 x 109/L. The baseline median palpable spleen length was 11.0 cm below the left costal margin; the median spleen volume [measured by magnetic resonance imaging (MRI) or computed tomography (CT)] was 2105 cm3 (range 610 to 9717 cm3).
Patients were treated with Omjjara 200 mg once daily or danazol 300 mg twice daily for 24 weeks, followed by open-label treatment with Omjjara. The two primary efficacy endpoints were percentage of patients with total symptom score (TSS) reduction of 50% or greater from baseline to week 24 (as measured by the Myelofibrosis Symptom Assessment Form [MFSAF] v4.0), and the percentage of patients who were transfusion independent (TI) at week 24 (defined as no transfusions and all haemoglobin values ≥ 8 g/dL in the 12 weeks prior to week 24).
The efficacy of Omjjara in the treatment of patients with primary or secondary myelofibrosis and anaemia was established based on a significantly higher percentage of patients treated with Omjjara compared to danazol, achieving a MFSAF v4.0 Total Symptom Score reduction of 50% or more at Week 24 compared with their own baseline score and by establishing non-inferiority of Omjjara with danazol in transfusion independence in the last 12 weeks of randomised treatment.
A key secondary endpoint measured the percentage of subjects with ≥ 35% reduction in spleen volume from baseline at week 24. At Week 24, a significantly higher percentage of patients treated with Omjjara achieved a spleen volume reduction by 35% or greater from baseline (Table 3).
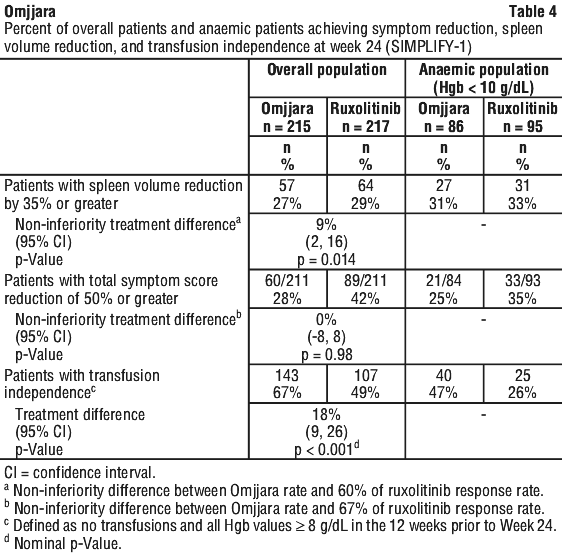
Patients were treated with Omjjara 200 mg or ruxolitinib adjusted dose twice daily for 24 weeks, followed by open-label treatment with Omjjara without tapering of ruxolitinib. The primary efficacy endpoint was percentage of patients with spleen volume response (reduction by 35% or greater) at Week 24; analyses were also conducted in a subset of patients with moderate to severe anaemia (Hgb < 10 g/dL) (Table 4). A similar percentage of patients treated with Omjjara or ruxolitinib achieved a spleen volume response in both populations. Other endpoints included TSS response and red blood cell transfusion requirements. Non-inferiority of Omjjara with ruxolitinib was not demonstrated for the first of the secondary endpoints, TSS reduction of 50% or greater.
5.2 Pharmacokinetic Properties
Absorption. Momelotinib is rapidly absorbed after oral administration with the maximal plasma concentration (Cmax) achieved within 3 hours post-dose, with plasma exposures increased in a less than dose proportional manner, especially at doses above 300 mg. At the dose of 200 mg once daily at steady state, the mean (% CV) momelotinib Cmax is 479 nanogram/mL (61%) and AUCtau is 3288 nanogram.h/mL (60%) in patients with myelofibrosis.
Following low-fat and high-fat meals in healthy volunteers, the Cmax of momelotinib was 38% and 28% higher, respectively, and the AUC was 16% and 28% higher, respectively, as compared with those under fasting conditions. These changes in exposure were not clinically meaningful.
Distribution. Plasma protein binding of momelotinib is approximately 91% in human. The mean apparent volume of distribution of momelotinib at steady-state was 984 L in patients with myelofibrosis receiving momelotinib 200 mg daily suggesting extensive tissue distribution.
Metabolism. Human metabolism of momelotinib is predominantly mediated by CYP enzymes with contributions in the following order: CYP3A4 (36%), CYP2C8 (19%), CYP2C19 (19%), CYP2C9 (17%), and CYP1A2 (9%). M21 is an active human metabolite that has approximately 40% of the pharmacological activity of the parent. M21 is formed by CYP followed by aldehyde oxidase metabolism of momelotinib. The mean M21 to momelotinib ratio for AUC ranged from 1.4 to 2.1.
Excretion. Following an oral dose of momelotinib 200 mg, the mean terminal half-life (t1/2) of momelotinib was 4 to 8 hours; the half-life of M21 is similar. The apparent total clearance (CL/F) of momelotinib was 103 L/h in patients with myelofibrosis.
Momelotinib is mainly eliminated through metabolism and then excreted to faeces. Following a single oral dose of [14C]-labelled momelotinib in healthy male subjects, 69% of radioactivity was excreted in the faeces (13% of dose as unchanged momelotinib), and 28% in the urine (< 1% of dose as unchanged momelotinib).
Special patient populations. Renal impairment. Momelotinib AUC decreased by 13% in subjects with moderate renal impairment (eGFR 30-59 mL/min/1.73 m2) and AUC decreased by 16% in subjects with severe renal impairment (eGFR 15-29 mL/min/1.73 m2) compared to subjects with normal renal function (eGFR ≥ 90 mL/min/1.73 m2). The AUC of the active metabolite, M21, increased by 20% and 41%, respectively, in subjects with moderate and severe renal impairment compared to subjects with normal renal function. There are no data in patients with end-stage renal disease (ESRD) receiving dialysis.
Hepatic impairment. Momelotinib AUC increased by 8% and 97% in subjects with moderate (Child-Pugh Class B) and severe (Child-Pugh Class C) hepatic impairment, respectively, compared to subjects with normal hepatic function (see Section 4.2 Dose and Method of Administration).
Age, gender, race, and bodyweight. Age, gender, race, or weight do not have a clinically meaningful effect on the pharmacokinetics of momelotinib based on a population pharmacokinetic analysis.
5.3 Preclinical Safety Data
Genotoxicity. Momelotinib was not mutagenic in a bacterial reverse mutation assay or clastogenic in an in vitro chromosomal aberration assay with human peripheral blood lymphocytes or in vivo in a rat bone marrow micronucleus assay.
Carcinogenicity. The carcinogenic potential of momelotinib was assessed in a 6-month rasH2 transgenic mouse study and a 2-year rat carcinogenicity study, both conducted by the oral route.
Momelotinib was not carcinogenic in transgenic mice up to the highest dose level tested (100 mg/kg/day, yielding exposure levels based on combined momelotinib and M21 [a major active human metabolite, minimally produced in mice] AUC 12-times the MRHD).
Increased incidence of testicular interstitial (Leydig) cell adenoma was noted in rats at a dose of 15 mg/kg/day (approximately 15-times the exposure based on combined momelotinib and M21 [minimally produced in rats] AUC at the MRHD). The increase in Leydig cell adenomas is considered to be a species-specific effect secondary to the induction of the prolactin signaling pathway, and, therefore, does not constitute a clinical carcinogenic risk.
6 Pharmaceutical Particulars
6.1 List of Excipients
Microcrystalline cellulose, lactose monohydrate, sodium starch glycolate (type A), magnesium stearate, colloidal anhydrous silica, propyl gallate, polyvinyl alcohol, macrogol 3350, titanium dioxide, purified talc, iron oxide yellow, iron oxide red.
6.2 Incompatibilities
Incompatibilities were not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C.
Store in the original bottle to protect from moisture. Do not remove the desiccant.
6.5 Nature and Contents of Container
White, high-density polyethylene (HDPE) bottles with child-resistant polypropylene cap and induction-sealed, aluminium faced liner. Each bottle contains 30 film-coated tablets, silica gel desiccant, and polyester coil packing material.
Not all strengths, dose forms, pack sizes, container types may be distributed in Australia.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Momelotinib dihydrochloride monohydrate is a light yellow to brown to reddish-brown solid, very slightly soluble to practically insoluble in water across the physiological pH range. The IUPAC chemical name is N-(cyanomethyl)-4-(2-{[4-(morpholin-4-yl) phenyl]amino}pyrimidin-4-yl)benzamide dihydrochloride hydrate.
Molecular formula: C23H22N6O2.2HCl.H2O.
C23H22N6O2 (free base).
Chemical structure.
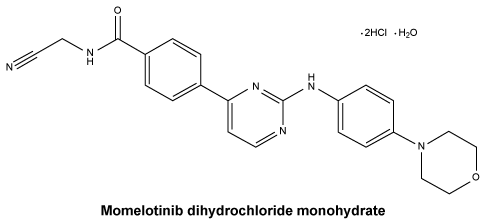
Momelotinib (free base) = 414.47.
CAS number. Momelotinib dihydrochloride monohydrate: 1841094-17-4.
Momelotinib (free base): 1056634-68-4.
7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.
Date of First Approval
18 December 2024
Date of Revision
03 February 2026
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.