Poteligeo
Brand Information
| Brand name | Poteligeo |
| Active ingredient | Mogamulizumab |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Poteligeo.
Summary CMI
POTELIGEO®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
▼ This medicine is new or being used differently. Please report side effects. See the full CMI for further details.
1. Why am I using POTELIGEO?
POTELIGEO contains the active ingredient mogamulizumab. POTELIGEO is used to treat adults with mycosis fungoides or Sézary syndrome, which are types of cancers called cutaneous T-cell lymphomas.
For more information, see Section 1. Why am I using POTELIGEO? in the full CMI.
2. What should I know before I use POTELIGEO?
Do not use it if you have ever had an allergic reaction to POTELIGEO or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines or are pregnant or plan to become pregnant or are breast-feeding.
For more information, see Section 2. What should I know before I use POTELIGEO? in the full CMI.
3. What if I am taking other medicines?
No studies have been performed to find out which medicines interfere with POTELIGEO and affect how it works.
For more information, see Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use POTELIGEO?
- The amount of POTELIGEO you will receive is calculated by your doctor based on your body weight.
- POTELIGEO will be given to you through a vein (intravenous infusion) over at least 60 minutes. To start with, the infusions will be given once a week for the first 5 doses, then once every 2 weeks.
More information can be found in Section 4. How do I use POTELIGEO? in the full CMI.
5. What should I know while using POTELIGEO?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using POTELIGEO? in the full CMI.
6. Are there any side effects?
Tell your doctor or nurse or get medical help immediately if you have any of the following symptoms after starting POTELIGEO:
- chills, nausea, vomiting, headache, wheezing, itching, flushing, rash, dizziness or feeling faint, difficulty breathing and fever, which may be signs of an infusion reaction.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
▼ This medicine is subject to additional monitoring. This will allow quick identification of new safety information. You can help by reporting any side effects you may get. You can report side effects to your doctor, or directly at www.tga.gov.au/reporting-problems.
POTELIGEO®
Active ingredient(s): Mogamulizumab
Consumer Medicine Information (CMI)
This leaflet provides important information about using POTELIGEO. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using POTELIGEO.
Where to find information in this leaflet:
1. Why am I using POTELIGEO?
2. What should I know before I use POTELIGEO?
3. What if I am taking other medicines?
4. How do I use POTELIGEO?
5. What should I know while using POTELIGEO?
6. Are there any side effects?
7. Product details
1. Why am I using POTELIGEO?
POTELIGEO contains the active substance mogamulizumab, which belongs to a group of medicines called monoclonal antibodies. Mogamulizumab targets a certain type of cancer cells which are then destroyed by the immune system (the body's defence).
This medicine is used to treat adults with mycosis fungoides or Sézary syndrome, which are types of cancers called cutaneous T-cell lymphomas. POTELIGEO will be prescribed only after you have tried another medicine given by mouth or by injection.
2. What should I know before I use POTELIGEO?
Warnings
Do not use POTELIGEO if:
- you are allergic to mogamulizumab, or any of the other ingredients listed at the end of this leaflet.
- Always check the ingredients to make sure you can use this medicine.
Check with your doctor if you:
- develop a skin reaction with this medicine. Fatal and life threatening skin reactions have occurred in some patients who have taken POTELIGEO.
- develop signs of infection including skin infections. This includes signs and symptoms such as fever, sweats, chills, flu-like symptoms, sore throat or skin sores or blisters that leak pus or fluid.
- develop an infusion reaction with this medicine (possible symptoms of an infusion reaction are listed in Section 6).
- have human immunodeficiency virus (HIV), herpes, cytomegalovirus (CMV), or hepatitis B or C infection, or other on-going infections.
- have had or plan to have a stem cell transplant, either using your own cells or a donor's.
- have had tumour lysis syndrome (a complication involving the destruction of cancer cells) after a previous treatment.
- have heart problems.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
Talk to your doctor if you are breast-feeding or intend to breastfeed.
The effects of POTELIGEO in pregnancy and breast-feeding are not known. Due to the effect of the medicine in your body, it may harm your baby if it is administered when you are pregnant or breast-feeding.
If you can get pregnant, you will need to use effective contraception during and for at least three months after receiving this treatment. If you are breastfeeding, you should discuss with your doctor whether you can breastfeed during or after treatment with POTELIGEO.
Men
You will need to use effective contraception during and for at least three months after receiving this treatment.
Children and adolescents
This medicine should not be used in children and adolescents below 18 years of age.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect POTELIGEO.
4. How do I use POTELIGEO?
How much to use
The amount of POTELIGEO you will receive is calculated by your doctor based on your body weight.
How POTELIGEO is given
POTELIGEO will be given to you through a vein (intravenous infusion) over at least 60 minutes. To start with, the infusions will be given once a week for the first 5 doses, then once every 2 weeks. You and your doctor will decide how long to stay on POTELIGEO. You may get serious side effects or the medicine may stop working.
It is recommended to take a medicine to treat fever and an anti-histamine (used for allergic reactions) before the first POTELIGEO infusion. If an infusion reaction (See Section 6 “Serious side effects”) occurs, these medicines are needed for subsequent POTELIGEO infusions.
If you are given too much (overdose)
As POTELIGEO is given to you under the supervision of your doctor it is unlikely that you will be given too much. However, if you experience any side effects after being given POTELIGEO, tell your doctor immediately.
5. What should I know while using POTELIGEO?
Things you should do
Tell the person giving you the infusion or get medical help straight away if you experience a reaction during or after any POTELIGEO infusion.
Tell your doctor immediately if you experience any of the serious side effects listed in Section 6 after starting POTELIGEO treatment.
Remind any doctor, dentist or pharmacist you visit that you are using POTELIGEO.
Things you should not do
Do not stop attending your appointments for your POTELIGEO infusion. You should advise your doctor immediately if you cannot attend the appointment to receive your infusion. It is important that you speak with your doctor to discuss re-scheduling your appointment as soon as possible.
Driving and using machines
Be careful before you drive or use any machines or tools until you know how POTELIGEO affects you.
POTELIGEO can affect your ability to drive and use machines. This medicine may cause tiredness after it has been given.
Looking after your medicine
- POTELIGEO must be stored in a refrigerator (2°C to 8°C) in the hospital or pharmacy.
- Do not freeze.
- Do not shake.
- Keep the vial in the outer carton in order to protect from light.
POTELIGEO contains sodium
This medicine contains less than 23 mg sodium per vial. Talk to your doctor or pharmacist about your sodium intake.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
Blood related:
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
Possible signs of inflammation of the large bowel (colitis):
The destruction of cancer cells and the body's reaction to it can very occasionally lead to a problem called tumour lysis syndrome.
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting of side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What POTELIGEO contains
| Active ingredient (main ingredient) | Mogamulizumab |
| Other ingredients (inactive ingredients) | The other excipients are citric acid monohydrate, glycine, polysorbate 80, sodium hydroxide, hydrochloric acid, and water for injections. See Section 5 “POTELIGEO contains sodium”. |
Do not use this medicine if you are allergic to any of these ingredients.
What POTELIGEO looks like and contents of the pack
POTELIGEO is a clear, colourless solution. The pack contains a glass vial containing 5 mL of concentrated solution for intravenous infusion (Aust R 330232).
Who distributes POTELIGEO
Kyowa Kirin Australia Pty Ltd
Level 7
68 York Street
Sydney, NSW 2000
This leaflet was prepared July 2025
Brand Information
| Brand name | Poteligeo |
| Active ingredient | Mogamulizumab |
| Schedule | S4 |
▼ This medicinal product is subject to additional monitoring in Australia. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse events at www.tga.gov.au/reporting-problems.
MIMS Revision Date: 01 April 2026
1 Name of Medicine
Mogamulizumab.
2 Qualitative and Quantitative Composition
Each vial of Poteligeo contains 20 mg of mogamulizumab, a recombinant defucosylated humanised IgG1 kappa monoclonal antibody produced in Chinese hamster ovary cells by recombinant DNA technology.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Concentrated solution for intravenous infusion. Clear to slightly opalescent, colourless solution.
4 Clinical Particulars
4.1 Therapeutic Indications
Poteligeo is indicated for the treatment of adult patients (≥ 18 years of age) with mycosis fungoides (MF) or Sézary syndrome (SS) who have received at least one prior systemic therapy.
4.2 Dose and Method of Administration
Treatment must be initiated and supervised by physicians experienced in the treatment of cancer and should only be administered by healthcare professionals in an environment where resuscitation equipment is available.
Dosage. The recommended dose is 1 mg/kg mogamulizumab administered as an intravenous infusion over at least 60 minutes. Administration is weekly on days 1, 8, 15 and 22 of the first 28-day cycle, followed by infusions every two weeks on Days 1 and 15 of each subsequent 28-day cycle until disease progression or unacceptable toxicity.
Poteligeo should be administered within 2 days of the scheduled day. If a dose is missed by more than 2 days, the next dose should be administered as soon as possible, after which the dosing schedule should be resumed with doses given based on the new scheduled days.
Pre-medication with anti-pyretic and anti-histamine is recommended for the first Poteligeo infusion. If an infusion reaction occurs, administer pre-medication for subsequent Poteligeo infusions.
Dose modification. Dermatologic reactions. Patients receiving mogamulizumab have experienced drug rash (drug eruption), some of which were severe and/or serious.
In the event of a rash (drug related) with severity of Grade 2 or 3 (moderate or severe), treatment with mogamulizumab must be interrupted and the rash should be treated appropriately until rash improves to Grade 1 or less (mild severity), at which time mogamulizumab treatment may be resumed.
Poteligeo should be permanently discontinued for a life-threatening (Grade 4) rash (see Section 4.4).
Infusion-related reactions. The infusion of Poteligeo should be temporarily interrupted for mild to severe (Grades 1-3) infusion-related reactions and symptoms treated. The infusion rate should be reduced by at least 50% when re-starting the infusion after symptoms resolve. If reaction recurs, discontinuing the infusion should be considered (see Section 4.4).
Poteligeo should be permanently discontinued for a life-threatening (Grade 4) infusion-related reaction (see Section 4.4).
Preparation for administration. Visually inspect the medicinal product for particulate matter and discolouration prior to administration. Poteligeo is a clear to slightly opalescent, colourless solution. Discard the vial if cloudiness, discolouration or particulates are observed.
Calculate the required volume of Poteligeo needed to prepare the infusion solution for the 1 mg/kg dosage based on patient weight. Aseptically withdraw the required volume of Poteligeo into the syringe and transfer into an infusion bag containing 9 mg per mL (0.9%) sodium chloride solution for injection. Mix diluted solution by gentle inversion. Do not shake. The final concentration of the diluted solution should be between 0.1 mg/mL to 3.0 mg/mL.
Product is for single use in one patient only. Discard any unused portion left in the vial in accordance with local requirements.
Administration. Poteligeo is for intravenous use. It should be administered by intravenous infusion only, over at least 60 minutes. See above recommendations in case of infusion-related reaction.
For instructions on the dilution of the medicinal product before administration:
The diluted solution is compatible with polyvinyl chloride (PVC) or polyolefin (PO) infusion bags.
Do not mix Poteligeo with, or administer as an infusion with, other medicinal products.
Poteligeo is intended for intravenous use only, and should not be administered subcutaneously, intramuscularly, as a bolus dose or by rapid intravenous administration.
Administer infusion solution over at least 60 minutes through an intravenous line containing a sterile, low protein binding 0.22 micron (or equivalent) in-line filter.
Special populations. Elderly (≥ 65 years of age). No dose adjustment is required in elderly patients (see Section 5.2).
Renal impairment. Based on a population pharmacokinetic analysis, no dose adjustment is recommended in patients with mild to severe renal impairment (see Section 5.2).
Hepatic impairment. Based on a population pharmacokinetic analysis, no dose adjustment is recommended in patients with mild or moderate hepatic impairment. Poteligeo has not been studied in patients with severe hepatic impairment (see Section 5.2).
4.3 Contraindications
Known hypersensitivity to mogamulizumab or to any of the excipients listed in Section 6.1 List of Excipients.
4.4 Special Warnings and Precautions for Use
Dermatologic reactions. Fatal and life-threatening skin adverse reactions, including Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), have occurred in recipients of mogamulizumab. Rash (drug eruption) is one of the most common adverse reactions associated with mogamulizumab. In Study 0761-010, 24% (44/184) of patients treated with mogamulizumab in the randomised phase of the study had an adverse reaction of drug eruption, with 4.3% of these cases being severe (Grade 3) and 95.7% of these cases being Grade 1 or 2. Of 528 patients treated with mogamulizumab in clinical trials, Grade 3 skin adverse reactions were reported in 3.6%, Grade 4 skin adverse reactions in < 1%, and SJS in < 1%.
The onset of drug eruption is variable, and the affected areas and appearance vary. In Study 0761-010, the median time to onset was 15 weeks, with 25% of cases occurring after 31 weeks. The more common presentations reported included papular or maculopapular rash, lichenoid, spongiotic or granulomatous dermatitis, and morbilliform rash. Other presentations included scaly plaques, pustular eruption, folliculitis, non-specific dermatitis, and psoriasiform dermatitis.
Monitor patients for rash throughout the treatment course. Management of dermatologic reactions includes topical corticosteroids and interruption or permanent cessation of mogamulizumab (see Section 4.2). Consider skin biopsy to help distinguish drug eruption from disease progression.
Discontinue mogamulizumab permanently for SJS or TEN or for any life-threatening (Grade 4) reaction. For possible SJS or TEN, interrupt mogamulizumab and do not restart unless SJS or TEN is ruled out and the cutaneous reaction has resolved to Grade 1 or less.
Infusion related reactions. Fatal and life-threatening infusion related reactions have been reported in patients treated with mogamulizumab. In Study 0761-010, infusion related reactions occurred in 33% (61/184) of patients treated with mogamulizumab in the randomised phase of the study, with 1.6% of these reactions being severe (Grade 3). In the crossover phase of the study, 37% (50/136) of the patients who switched to mogamulizumab treatment experienced infusion related reactions, with 4.4% being severe (Grade 3). Most reactions (approximately 90%) occur during or shortly after the first cycle of infusion (first four administrations). Infusion related reactions can also occur with subsequent infusions. The most commonly reported signs include chills, nausea, fever, tachycardia, rigors, headache, and vomiting.
Consider premedication (such as anti-histamines and paracetamol) for the first infusion of mogamulizumab in all patients. Whether premedication reduces the risk or severity of these reactions is not established. In Study 0761-010, infusion related reactions occurred in 42% of patients without premedication and 32% of patients with premedication. Monitor patients closely for signs and symptoms of infusion related reactions and interrupt the infusion for any grade reaction and treat promptly (see Section 4.2).
Infections. Fatal and life-threatening infections have occurred in patients treated with mogamulizumab, including sepsis, pneumonia, and skin infection. In Study 0761-010, 17% (32/184) of patients randomised to mogamulizumab had Grade 3 or higher infection or an infection-related serious adverse reaction. Monitor patients for signs and symptoms of infection and treat promptly.
Patients should be tested for hepatitis B infection before initiating treatment with mogamulizumab. For patients who test positive for current/previous hepatitis B infection, consultation with a physician with expertise in the treatment of hepatitis B is recommended for advice concerning appropriate measures against hepatitis B reactivation.
Complications of allogeneic hematopoietic stem cell transplantation (HSCT) after mogamulizumab. Increased risks of transplant complications have been reported in patients who receive allogeneic HSCT after mogamulizumab including severe (Grade 3 or 4) acute graft-versus-host disease (GVHD), steroid-refractory GVHD, and transplant-related death. Among recipients of pre-transplantation mogamulizumab, a higher risk of transplant complications has been reported if mogamulizumab is given within a shorter time frame (approximately 50 days) before HSCT. Follow patients closely for early evidence of transplant-related complications.
Tumour lysis syndrome. Tumour lysis syndrome (TLS) has been observed in patients receiving mogamulizumab. TLS was observed most frequently during the first month of treatment. Patients with rapidly proliferating tumour and high tumour burden are at risk of TLS. Patients should be monitored closely by appropriate laboratory and clinical tests for electrolyte status, hydration and renal function, particularly in the first month of treatment, and managed according to best medical practice. Management of TLS may include aggressive hydration, correction of electrolyte abnormalities, anti-hyperuricaemic therapy, and supportive care.
Cardiac disorders. One case of acute myocardial infarction has been observed in a clinical trial patient with MF / SS receiving mogamulizumab. In clinical trial patients with other T-cell lymphomas there have been reports of stress cardiomyopathy (one case) and acute myocardial infarction (one case). The subjects had a medical history including various risk factors. Patients who have risk factors associated with cardiac disease should be monitored and appropriate precautions taken.
Large cell transformation (LCT). There are limited data available on patients with LCT.
Other. Mogamulizumab should not be administered subcutaneously or intramuscularly, by rapid intravenous administration, or as an intravenous bolus.
Use in the elderly. Poteligeo may be administered to patients aged 65 years and over.
Paediatric use. The safety and efficacy of Poteligeo in children and adolescents aged below 18 years have not been established. No data are available.
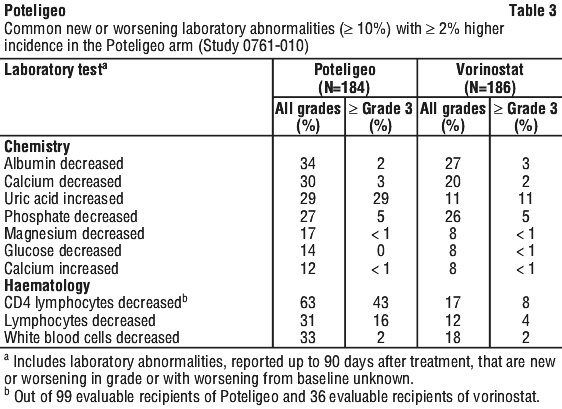
Effects on laboratory tests. Poteligeo may cause increased alanine aminotransferase, increased aspartate aminotransferase, increased blood alkaline phosphatase or decreased lymphocyte count. Common or worsening laboratory abnormalities are summarised, see Section 4.8, Table 3.
4.5 Interactions with Other Medicines and Other Forms of Interactions
No interaction studies have been performed.
4.6 Fertility, Pregnancy and Lactation
Women of childbearing potential/ contraception in males and females. Women of childbearing potential and males of reproductive potential should use effective contraception during treatment with Poteligeo and for at least 3 months after treatment.
Effects on fertility. There are no clinical data available on the effect of Poteligeo on human fertility. No specific studies in animals have been performed to evaluate the effect of mogamulizumab on fertility. No adverse effects on male and female reproductive organs were observed in repeat-dose toxicity studies in cynomolgus monkeys at intravenous doses up to 40 mg/kg/week for 26 weeks (yielding almost 80 times the systemic exposure in patients, based on AUC).
Use in pregnancy. (Category C)
There are no data from the use of Poteligeo in pregnant women.
In a study in pregnant cynomolgus monkeys, mogamulizumab did not cause malformations, embryofetal lethality or growth retardation with administration at 40 mg/kg/week IV (yielding 27 times the systemic exposure in patients, based on AUC). However, mogamulizumab was shown to cross the placental barrier, and this resulted in pharmacological activity in the foetuses, evident as a decrease in CCR4-expressing lymphocytes. Poteligeo is not recommended during pregnancy unless the potential benefit for the mother outweighs the potential risk to the foetus.
Use in lactation. It is unknown whether mogamulizumab is excreted in human milk. Human IgGs are known to be excreted in breast milk. Consequently, a risk to the breast-fed child cannot be excluded. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for Poteligeo and any potential adverse effects on the breastfed child from Poteligeo.
4.7 Effects on Ability to Drive and Use Machines
Mogamulizumab has minor influence on the ability to drive and use machines. Fatigue may occur following administration of mogamulizumab (see Section 4.8).
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile. The most frequently reported serious adverse reactions were pneumonia, pyrexia, infusion related reaction and cellulitis.
The most frequently reported adverse reactions were infusion-related reaction and rash (drug eruption); most of these reactions were non-serious and Grades 1 or 2.
Severe adverse reactions included Grade 4 respiratory failure (1.1%) and Grade 5 reactions were polymyositis and sepsis (0.5% each).
Tabulated list of adverse reactions. The adverse reactions are presented by system organ class and frequency categories, defined using the following convention: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000), not known (cannot be estimated from the available data). Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness. See Table 1.

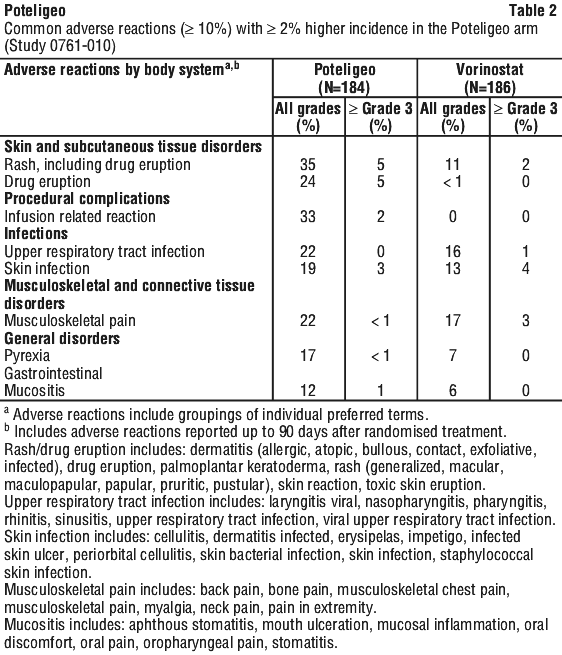
Infusion-related reactions. Infusion-related reactions have been observed in 33% of patients treated with Poteligeo. The majority of treatment-related infusion-related reactions were Grade 1 or 2 and occurred during or shortly after the first infusion. Severe reactions (Grade 3) were experienced by 1.6% of the patients in the randomised phase of the study and 4.4% of patients in the crossover phase of the study.
The incidence of infusion related reactions was highest after the first infusion (28.8% of patients), reducing to ≤ 3.8% of patients after two or more infusions.
Infusion interruptions occurred in approximately 6% of patients, most of which (approximately 90%) occurred within the first cycle of treatment with mogamulizumab.
Less than 1% of patients treated in Study 0761-010 discontinued treatment due to infusion-related reactions.
Serious infections. Patients with MF or SS are at increased risk of serious infection due to the disruption of dermal integrity caused by cutaneous disease, as well as the immunosuppressive effects of extracutaneous disease, and treatment with mogamulizumab may increase that risk. Serious infections, including sepsis, pneumonia and skin infections, were experienced by 14.3% of subjects receiving mogamulizumab. The latency to event onset following the first dose varied considerably. The majority of patients recovered from infection. In the clinical trial (0761-010), there were 2 reports of respiratory failure with fatal outcome in patients with severe pneumonia occurring more than 9 months after starting treatment with mogamulizumab.
Immune-mediated events. Fatal and life-threatening immune-mediated complications have been reported in recipients of mogamulizumab. Grade 3 or higher immune-mediated or possibly immune-mediated reactions have included myositis, myocarditis, polymyositis, hepatitis, pneumonitis, and a variant of Guillain-Barré syndrome. Use of systemic immunosuppressants for immune-mediated reactions was reported in 1.9% (6/319) of recipients of mogamulizumab in Study 0761-010, including for a case of Grade 2 polymyalgia rheumatica. New-onset hypothyroidism (Grade 1 or 2) was reported in 1.3% of patients and managed with observation or levothyroxine. Interrupt or permanently discontinue mogamulizumab as appropriate for suspected immune-mediated adverse reactions. Consider the benefit/risk of mogamulizumab in patients with a history of autoimmune disease.
Immunogenicity. As with all therapeutic proteins, there is a potential for immunogenicity. A small percentage of patients receiving Poteligeo tested positive for treatment emergent (treatment induced or treatment boosted) anti mogamulizumab antibodies. There were no positive neutralising antibody responses.
Gastrointestinal disorders. Colitis was mainly characterised by watery diarrhoea, in some cases excessive.
Safety post last dose. Of the 320 subjects exposed to mogamulizumab in Study 0761-010, 21 (6.6%), experienced at least one serious adverse drug reaction (SADR) that occurred within 90 days from the date of last study drug administration.
Of these, SADRs that were reported in more than one patient were coded under the SOCs Infections and infestations (7 [2.2%] patients), General disorders and administration site conditions (5 [1.6%] patients), Respiratory, thoracic and mediastinal disorders (4 [1.3%] patients), Musculoskeletal and connective tissue disorders (3 [0.9%] patients), Hepatobiliary disorders (2 [0.6%] patients), and Injury, poisoning and procedural complications (2 [0.6%] patients). All remaining SOCs reported SADRs in one patient (0.3%).
The safety profile observed in the 90 days following the last dose of mogamulizumab is consistent with the safety profile observed during the study treatment period.
Elderly population. The safety profile in elderly patients (≥ 65 years) was generally consistent with that of adult patients, except for dermatologic reactions and infusion related reactions which were seen more often in older subjects.
Reporting suspected adverse effects. Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions www.tga.gov.au/reporting-problems.
4.9 Overdose
There is no information on overdose with mogamulizumab. In case of overdose, the patient, including their vital signs, should be closely monitored (for at least 1 hour) and supportive treatment should be administered if required.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action. Mogamulizumab is a defucosylated, humanised IgG1 kappa monoclonal antibody that selectively binds to CCR4, a G protein-coupled receptor for CC chemokines that is involved in the trafficking of lymphocytes to various organs including the skin. CCR4 is expressed on the surface of some T cell malignancies, such as MF and SS as well as Type 2 T helper (Th2) T cells and regulatory T cells (Tregs). Binding of mogamulizumab to CCR4 induces antibody-dependent cellular cytotoxicity (ADCC), resulting in the depletion of target cells.
Clinical trials. The efficacy of mogamulizumab in the treatment of patients with mycosis fungoides (MF) or Sézary syndrome (SS) was established in a Phase 3, multicentre, open-label, study (0761-010) of 372 adult patients randomised 1:1 to treatment with either mogamulizumab or vorinostat. Each arm enrolled 186 patients. Mogamulizumab infusion was administered at a dose of 1 mg/kg once weekly for the first 28-day cycle (on days 1, 8, 15 and 22), and on days 1 and 15 of subsequent 28-day cycles. Vorinostat was administered at a starting dose of 400 mg orally, once daily beginning on Day 1 for 28-day cycles. Vorinostat patients with disease progression or unacceptable toxicities were permitted to cross over to mogamulizumab therapy. Crossover patients received up to 46 months of mogamulizumab therapy, as of December 2016 data cut. Treatment with mogamulizumab continued until disease progression or unacceptable toxicity. The trial excluded patients with active autoimmune diseases, central nervous system metastasis, and medical conditions that required systemic corticosteroids or other immunosuppressive medicinal products, or an active infection requiring therapy, including HIV, or hepatitis B or C. Patients with ECOG performance status ≥ 2 were also excluded. At study baseline, 38% had stage IB-II disease, 10% stage III, 52% stage IV. This study included patients regardless of their baseline level of CCR4 expression in skin biopsy.
The primary efficacy endpoint was progression-free survival (PFS) based on investigator assessment using a global composite response criteria that took into account all potentially affected disease compartments (skin, blood, lymph nodes and viscera). Response in skin and blood was evaluated every 4 weeks. Response in lymph nodes and viscera was evaluated at 4 weeks, then every 8 weeks in the first year, and then every 16 weeks thereafter.
All patients had a histologically confirmed diagnosis of mycosis fungoides (MF), 56.5%, 53.2%, or Sézary syndrome (SS), 43.5%, 46.8%, in the mogamulizumab and vorinostat groups, respectively, and had received at least one prior systemic therapy. The most common prior systemic therapies used by subjects in Europe were bexarotene (70%), interferon (59%), methotrexate (49%), extracorporeal photopheresis (ECP) (31%) and gemcitabine/gemcitabine regimens (28%).
The median duration of exposure with mogamulizumab was 5.6 months (range: < 1 to 45.3 months). 56% of patients received mogamulizumab for at least 6 cycles, and 25% of patients received mogamulizumab for at least 12 cycles.
Patients were a median age of 64 years at the time of screening (range 25 to 101 years), 49.5% were 65 years or older, and 58.1% were male.
CCR4 expression was assessed retrospectively on pre-treatment skin biopsies (formalin fixed paraffin embedded) using immunohistochemistry. In the mogamulizumab arm, baseline CCR4 expression levels were available in 75% of patients (N=140). CCR4 was detected on ≥ 1% of lymphocytes in 100% of patients, and 96% (134/140) had CCR4 detected on ≥ 10% of skin lymphocytes.
Of the patients randomised to vorinostat, 136 patients (73.1%) crossed over to mogamulizumab during the study. Reasons for crossover to mogamulizumab were disease progression (109 patients) and treatment intolerance (27 patients). The number of infusions of mogamulizumab administered to crossover patients ranged from 1 to 94 (up to 46 months of treatment) as of the December 2016 data cut.
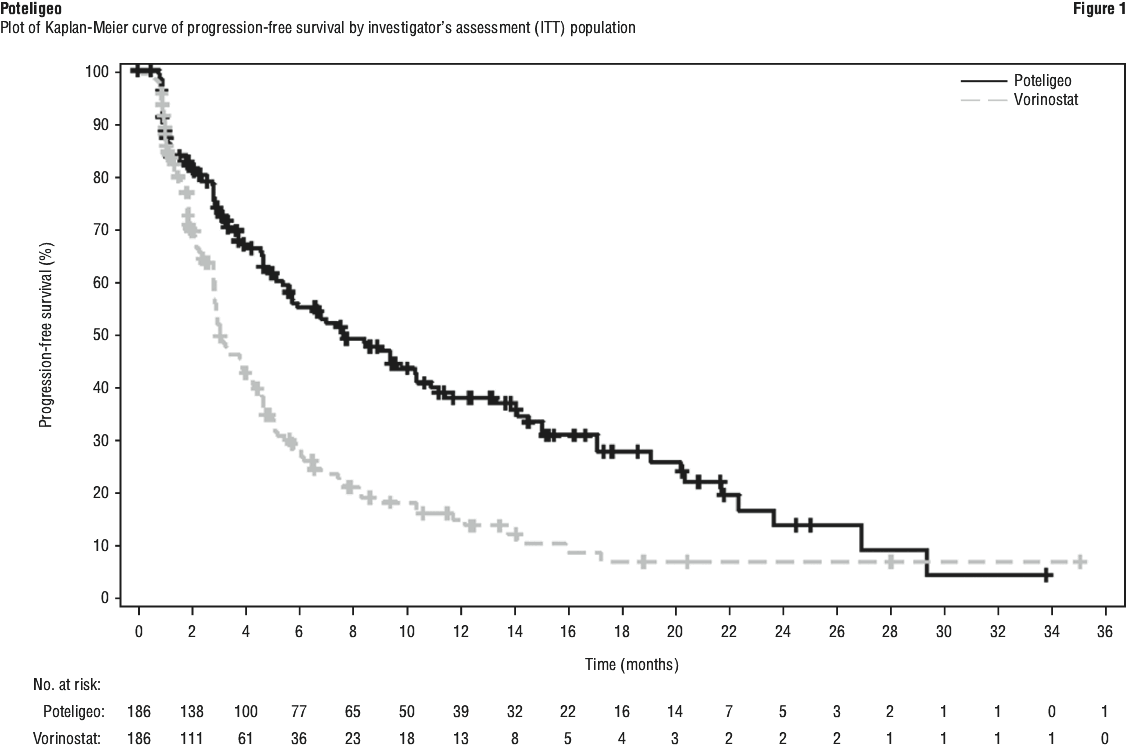
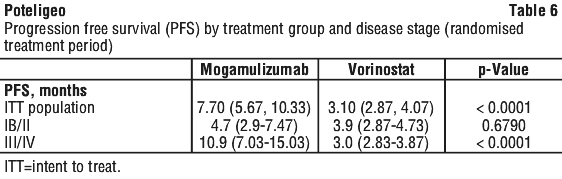
At 6, 12, 18 and 24 months after the start of randomised treatment, the percent of subjects alive without disease progression was higher for mogamulizumab (55.3%, 38.3%, 28.0%, and 14.1%, respectively) compared to vorinostat (28.8%, 15.3%, 7.2%, and 7.2%, respectively). Median PFS for the mogamulizumab group was 7.70 months (95% CI: 5.67, 10.33) and 3.10 months (95% CI: 2.87, 4.07) for the vorinostat group with resultant hazard ration of 0.53 (95% CI: 0.41, 0.69), p < 0.0001 (2-sided, stratified log rank test).
The Kaplan-Meier curve for PFS is shown Figure 1.
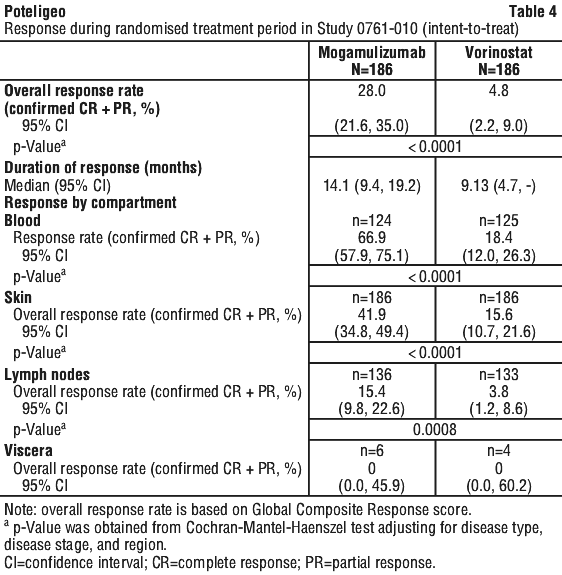
Overall response was reported as a composite score from measures in each compartment, and a response had to be demonstrated at two successive overall disease assessments (at least 8 weeks apart during the first year and 16 weeks apart thereafter) in order to be confirmed. Patients were included in the analysis for a specific compartment if they had presence of disease in that compartment at baseline, or had any post-baseline response assessment for that compartment.
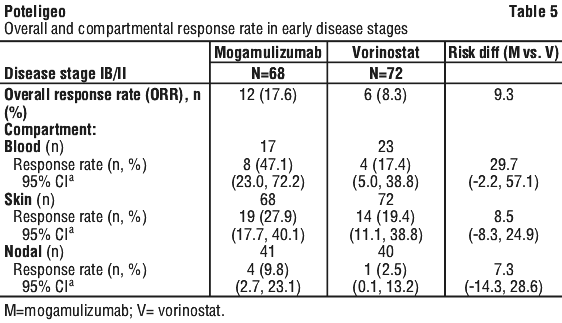
Table 4 summarises ORR and DOR, and response by compartment. The study demonstrated statistically significant improvements in ORR and response by compartment in the blood, skin, and lymph nodes as compared to vorinostat. Response in the viscera could not be evaluated due to limited efficacy data in subjects with visceral involvement; the benefit-risk of mogamulizumab in subjects with visceral involvement is currently undetermined due to lack of data.
There are limited efficacy data in patients with low (< 10%) CCR4 expression in the skin. In Study 0761-010 there were 10/290 evaluable patients with CCR4 expression < 10%, of which 6 were randomised to mogamulizumab, and 4 were randomised to vorinostat and subsequently crossed over to mogamulizumab. No confirmed responses were observed in these 10 subjects with low (< 10%) CCR4 expression. Compartmental responses were seen in 3 of 10 evaluable subjects treated with mogamulizumab in the randomised or cross over phase.
Patients with stage IB/II disease treated with mogamulizumab had confirmed ORR of 17.6% compared to 8.3% for vorinostat, and compartment level (blood, skin, lymph node) response rates that were higher than those for vorinostat treated patients (Table 5). Overall, the median period of progression free survival for stage IB/II subjects treated with mogamulizumab was 4.7 months compared to 3.9 months for vorinostat-treated patients (Table 6). In patients with stage IB/II disease, given the limited number of subjects with a response and immaturity of the data, no conclusion on duration of response can be made.
Time to compartment level response in stage IB/II patients was approximately 3 months, which is consistent with time to response for the ITT population overall (approximately 3 months). If a compartment level response or overall response is not observed after 3 months of treatment, discontinuation of treatment should be considered.
5.2 Pharmacokinetic Properties
The pharmacokinetics (PK) of mogamulizumab was evaluated in adult patients with T-cell leukaemia-lymphoma (ATL) and CTCL over a dose range of 0.01 to 1 mg/kg administered as multiple doses of mogamulizumab every week or every 2 weeks, and included the recommended 1.0 mg/kg dose and regimen (days 1, 8, 15 and 22 for the first 28-day cycle and on Days 1 and 15 for subsequent 28-day cycles). The population PK analysis included 444 patients receiving mogamulizumab in six clinical trials. The exposure to mogamulizumab increased proportionally with dose over the dose range of 0.1 to 1.0 mg/kg.
Absorption. Mogamulizumab is dosed via intravenous route and therefore is immediately and completely bioavailable.
Distribution. Based on a population PK analysis, the geometric mean [% coefficient of variation (CV%)] central volume of distribution (Vc) was 3.57 L (20.1%).
Metabolism. The metabolic pathway of mogamulizumab has not been characterised. Mogamulizumab is expected to be degraded into small peptides and amino acids via catabolic pathways in the same manner as endogenous IgG.
Excretion. Based on a population PK analysis, the geometric mean (% coefficient of variation [CV%]) clearance (CL) is 12.0 mL/h (83.7%) and geometric mean elimination half-life (t1/2) is 17 days (65.5%).
Linearity and accumulation. Mogamulizumab exhibits linear PK from the dose in a dose range of 0.01 mg/kg to 1 mg/kg. Based on a population PK analysis, the steady-state concentrations of mogamulizumab were reached after 12 weeks of repeated dosing when administered using the recommended regimen, and systemic accumulation was 1.7-fold. On a power model analysis, no deviation from dose proportionality was evident.
Renal impairment. The effect of renal impairment on the clearance of mogamulizumab was evaluated by a population PK analysis in patients with mild (creatinine clearance [CrCL] between 60 and 89; n=157), moderate (CrCL between 59 and 30; n=80), or severe renal impairment (CrCL less than 30 mL/min; n=2). No clinically important differences in the clearance of mogamulizumab were found between patients with mild to severe renal impairment and patients with normal renal function.
Hepatic impairment. The effect of hepatic impairment on the clearance of mogamulizumab was evaluated by a population PK analysis in patients with mild hepatic impairment (total bilirubin [TB] less than or equal to the upper limit of normal [ULN] and AST greater than ULN or TB less than 1 to 1.5 times ULN and any AST; n=80) or moderate (TB greater than 1.5 to 3 times ULN and any AST; n=3) hepatic impairment. No clinically important differences in the clearance of mogamulizumab were found between patients with mild to moderate hepatic impairment and patients with normal hepatic function. Mogamulizumab has not been studied in patients with severe hepatic impairment (TB greater than 3 times ULN and any AST).
Other special populations. The effects of various covariates on the PKs of mogamulizumab were assessed in population PK analyses. The following factors had no clinically important effect on the CL of mogamulizumab: age (range: 22 to 101 years), sex, ethnicity (other than Japanese, limited data are available in other ethnic populations), renal impairment, mild or moderate hepatic impairment, disease subtype (mycosis fungoides (MF) or Sézary syndrome (SS)), degree of CCR4 expression or ECOG status, although it should be noted that patients with ECOG PS ≥ 2 were excluded from the clinical trials.
Pharmacokinetic/pharmacodynamic relationship(s). Efficacy. Exposure-Response analysis indicated that efficacy was not correlated with mogamulizumab exposure in the pivotal study. Efficacy, as measured by improvement in PFS based on investigator assessment, was not associated with increasing mogamulizumab exposure.
5.3 Preclinical Safety Data
Genotoxicity. No studies have been conducted to assess the genotoxic potential of mogamulizumab.
As a large protein molecule, mogamulizumab is not expected to interact with DNA or other chromosomal material.
Carcinogenicity. No studies have been conducted to assess the carcinogenic potential of mogamulizumab.
6 Pharmaceutical Particulars
6.1 List of Excipients
Citric acid monohydrate, glycine, polysorbate 80, sodium hydroxide (for pH adjustment), hydrochloric acid (for pH adjustment), water for injections.
This medicinal product contains less than 1 mmol sodium per dose, that is to say essentially 'sodium free'.
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products. Mogamulizumab should not be infused concomitantly in the same intravenous line with other medicinal products.
6.3 Shelf Life
Unopened vial. 3 years.
After opening. Poteligeo does not contain a preservative. Once opened, the medicinal product should be diluted and infused immediately (see Section 4.2).
After preparation of infusion. Chemical and physical in-use stability has been demonstrated for 24 hours at room temperature (at 25°C) under ambient room light. These time limits include storage of the infusion solution in the infusion bag through the duration of infusion. From a microbiological point of view, the product must be used immediately. If not used immediately, in-use storage times and conditions prior to use are the responsibility of the user and must not be longer than a total of 4 hours at 2°C - 8°C provided that dilution has taken place under controlled and validated aseptic conditions.
6.4 Special Precautions for Storage
Must be stored in a refrigerator (2°C to 8°C).
Do not freeze.
Do not shake.
Keep the vial in the outer carton in order to protect from light.
For storage conditions after dilution of the medicinal product, see Section 6.3.
6.5 Nature and Contents of Container
5 mL solution in a 10 mL glass vial (type I glass) with a rubber stopper, an aluminium seal and a polypropylene flip-off cap.
Pack of one vial.
6.6 Special Precautions for Disposal
Product is for single use only. Discard any residue.
In Australia, any unused medicine or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
Chemical structure.
7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.
Date of First Approval
05 February 2021
Date of Revision
08 August 2025
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.