Praxbind
Brand Information
| Brand name | Praxbind |
| Active ingredient | Idarucizumab |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Praxbind.
Summary CMI
Praxbind®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I being given Praxbind?
Praxbind contains the active ingredient idarucizumab, which is a reversal agent specific for Pradaxa (dabigatran etexilate), a blood thinner medicine that helps prevent blood clots. Praxbind is used to rapidly trap dabigatran in order to inactivate its effect.
For more information, see Section 1. Why am I being given Praxbind? in the full CMI.
2. What should I know before I am given Praxbind?
Do not use if you have ever had an allergic reaction to idarucizumab or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I am given Praxbind? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with Praxbind and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How will I be given Praxbind?
A doctor or nurse will give you Praxbind by injection or infusion into a vein. It is not for self-administration.
More instructions can be found in Section 4. How will I be given Praxbind? in the full CMI.
5. What should I know after being given Praxbind?
| Things you should do |
|
For more information, see Section 5. What should I know after being given Praxbind? in the full CMI.
6. Are there any side effects?
Tell your doctor or nurse if you notice any of the following and they worry you: headache, back pain, fever, wheezing, rash and itchy skin.
Tell your doctor or nurse as soon as possible if you experience any side effects during or after treatment with Praxbind, so that these may be properly treated.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
Praxbind®
Active ingredient(s): idarucizumab
Consumer Medicine Information (CMI)
This leaflet provides important information about using Praxbind. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about being given Praxbind.
Where to find information in this leaflet:
1. Why am I being given Praxbind?
2. What should I know before I am given Praxbind?
3. What if I am taking other medicines?
4. How will I be given Praxbind?
5. What should I know after being given Praxbind?
6. Are there any side effects?
7. Product details
1. Why am I being given Praxbind?
Praxbind contains the active ingredient idarucizumab. Praxbind is a reversal agent specific for Pradaxa (dabigatran etexilate), a blood thinner medicine that blocks a substance in the body, which is involved in blood clot formation. Praxbind is used to rapidly trap dabigatran in order to inactivate its effect.
Praxbind is used in emergency situations where your doctor decides that rapid inactivation of the effect of Pradaxa (dabigatran etexilate) is required such as:
- for emergency surgery/urgent procedures
- in life-threatening or uncontrolled bleeding.
This medicine will only remove dabigatran from your body. It will not remove other medicines used to prevent the formation of blood clots.
After dabigatran has been removed from your body, you are not protected from the formation of blood clots. Your doctor will continue treating you with medicines used to prevent the formation of blood clots as soon as your medical condition allows.
Ask your doctor if you have any questions about why this medicine is being given to you.
Your doctor may have prescribed it for another reason.
2. What should I know before I am given Praxbind?
Warnings
You must not be given Praxbind if:
- You are allergic to idarucizumab, or any of the ingredients listed at the end of this leaflet.
Always check the ingredients to make sure you can use this medicine.
If you are not sure whether you should be given this medicine, talk to your doctor.
Check with your doctor if you:
- have allergies to any other medicines, foods, preservatives or dyes
- have any other medical conditions
- take any medicines for any other condition
- have a genetic disease called hereditary fructose intolerance. In this case, the substance sorbitol contained in Praxbind may cause serious adverse reactions.
- are on a sodium restricted diet. Praxbind contains 50 mg sodium per dose.
Your doctor will take this into account before treating you with Praxbind.
If you are uncertain as to whether you have, or have had, any of these conditions you should raise those concerns with your doctor.
If you have not told your doctor about any of the above, tell him/her before you are given Praxbind.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
Your doctor can discuss with you the risks and benefits involved.
Children and adolescents
There is no information on the use of Praxbind in children.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
This medicine has been designed to only bind to dabigatran. It is unlikely that other medicines will influence the effect of Praxbind or that Praxbind will influence the effect of other medicines.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect Praxbind.
4. How will I be given Praxbind?
How much Praxbind will I be given?
- The recommended dose of Praxbind is 5 g (2 vials of 50 mL).
- In rare cases you may still have too much dabigatran in your blood after a first dose of Praxbind and your doctor may decide to give you a second 5 g dose in specific situations.
How is Praxbind given?
- Your doctor or nurse will give you this medicine by injection or infusion into a vein.
- After you have received Praxbind, your doctor will decide on the continuation of your treatment to prevent blood clot formation. Pradaxa can be given again 24 hours after Praxbind administration.
If you are given too much Praxbind
As Praxbind is given to you under the supervision of your doctor, it is very unlikely that you will receive too much.
5. What should I know after being given Praxbind?
Things you should do
Call your doctor straight away if you notice any of the following:
- long or excessive bleeding
- exceptional weakness
- tiredness, headaches, dizziness and looking pale (signs of anaemia)
- chest pain or being short of breath
- red or dark brown urine
- red or black bowel motions.
These are signs or symptoms of bleeding. You may need urgent medical attention.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Side effects
| Side effects | What to do |
Signs of an allergic reaction
| Tell your doctor or nurse as soon as possible if you experience any side effects during or after treatment with Praxbind, so that these may be properly treated. |
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What Praxbind contains
| Active ingredient (main ingredient) | Each 50 mL vial of Praxbind contains 2.5 g of idarucizumab |
| Other ingredients (inactive ingredients) |
|
Do not take this medicine if you are allergic to any of these ingredients.
What Praxbind looks like
Praxbind (AUST R 237761) is a clear to slightly opalescent, colourless to slightly yellow solution. Praxbind is supplied in a 50 mL glass vial, closed with a rubber stopper and secured with an aluminium flip-off cap.
Praxbind is available in packs of 2 vials.
Who distributes Praxbind
Praxbind is supplied in Australia by:
Boehringer Ingelheim Pty Limited
ABN 52 000 452 308
Sydney NSW
www.boehringer-ingelheim.com.au
® Praxbind is a registered trade mark of Boehringer Ingelheim.
© Boehringer Ingelheim Pty Limited 2026
This leaflet was prepared in April 2026.
Brand Information
| Brand name | Praxbind |
| Active ingredient | Idarucizumab |
| Schedule | S4 |
MIMS Revision Date: 01 March 2022
1 Name of Medicine
Idarucizumab, rch.
2 Qualitative and Quantitative Composition
Each 50 mL vial of Praxbind solution for injection/infusion contains 2.5 g of idarucizumab (50 mg/mL).
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Praxbind 50 mg/mL solution for injection/infusion is a clear to slightly opalescent, colourless to slightly yellow solution.
4 Clinical Particulars
4.1 Therapeutic Indications
Praxbind is a specific reversal agent for dabigatran and is indicated in patients treated with dabigatran etexilate (Pradaxa) when rapid reversal of the anticoagulant effects of dabigatran is required:
for emergency surgery/urgent procedures;
in life threatening or uncontrolled bleeding.
4.2 Dose and Method of Administration
Dosage. The recommended dose of Praxbind is 5 g (2 x 2.5 g/50 mL).
Praxbind (2 x 2.5 g/50 mL) is administered intravenously, as two consecutive infusions over 5 to 10 minutes each (by hanging vials) or as a bolus injection (inject both vials consecutively via syringe).
See manufacturer's product information for diagrams.
In a limited number of patients, recurrence of plasma concentrations of unbound dabigatran and concomitant prolongation of clotting tests has occurred up to 24 hours after administration of idarucizumab (see Section 5.1 Pharmacodynamic Properties).
Administration of a second 5 g dose of Praxbind may be considered in the following situations:
recurrence of clinically relevant bleeding together with prolonged clotting times; or
patients require a second emergency surgery/urgent procedure and have prolonged clotting times.
Relevant coagulation parameters are activated partial thromboplastin time (aPTT), diluted thrombin time (dTT) or ecarin clotting time (ECT).
The safety and efficacy of repeat treatment with Praxbind have not been established.
Restarting antithrombotic therapy. Patients being treated with Pradaxa have underlying disease states that predispose them to thromboembolic events. Reversing Pradaxa exposes patients to the thrombotic risk of their underlying disease. To reduce this risk, resumption of anticoagulant therapy should be considered as soon as medically appropriate. Idarucizumab is a specific reversal agent for dabigatran, with no impact on the effect of other anticoagulant or antithrombotic therapies.
Pradaxa treatment can be initiated 24 hours after administration of Praxbind (refer to dosing, see Section 4.4 Special Warnings and Precautions for Use, Use in renal impairment).
Method of administration. Instructions for use / handling. Parenteral drug products should be inspected visually for particulate matter and discolouration prior to administration.
Praxbind must not be mixed with other medicinal products. A pre-existing intravenous line may be used for administration of Praxbind. The line must be flushed with sterile sodium chloride 9 mg/mL (0.9%) solution prior to and at the end of infusion. No other infusion should be administered in parallel via the same intravenous access.
Prior to use, the unopened vial may be kept at room temperature (up to 30°C) for up to 48 hours, if stored in the original package in order to protect from light. Once solution has been removed from the vial, chemical and physical in-use stability of idarucizumab has been demonstrated for up to 6 hours at room temperature. The solution should not be exposed to light for more than 6 hours.
Praxbind does not contain preservatives. Praxbind is for single use in one patient only. Discard any residue.
No incompatibilities between Praxbind and polyvinyl chloride, polyethylene or polyurethane infusion sets or polypropylene syringes have been observed.
4.3 Contraindications
None.
4.4 Special Warnings and Precautions for Use
Safety and efficacy in patients has been evaluated in 503 patients in a prospective, open label, nonrandomised, uncontrolled study (RE-VERSE AD) (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Idarucizumab binds specifically to dabigatran and reverses its anticoagulant effect. It will not reverse the effects of other anticoagulants (see Section 5.1 Pharmacodynamic Properties).
Praxbind treatment can be used in conjunction with standard supportive measures, which should be considered as medically appropriate.
Traceability. In order to improve traceability of biological medicinal products, the trade name and the batch number of the administered product should be clearly recorded in the patient file.
Thromboembolic events. Patients being treated with dabigatran have underlying disease states that predispose them to thromboembolic events. Reversal of dabigatran therapy exposes patients to the thrombotic risk of their underlying disease. To reduce this risk, resumption of anticoagulant therapy should be considered as soon as medically appropriate (see Section 4.2 Dose and Method of Administration).
Hypersensitivity. The risk of using Praxbind in patients with known hypersensitivity (e.g. anaphylactoid reaction) to idarucizumab or to any of the excipients needs to be weighed cautiously against the potential benefit of such an emergency treatment. If an anaphylactic reaction or other serious allergic reaction occurs, administration of Praxbind should be discontinued immediately and appropriate therapy initiated.
Hereditary fructose intolerance. The recommended dose of Praxbind contains 4 g sorbitol as an excipient. In patients with hereditary fructose intolerance, parenteral administration of sorbitol has been associated with reports of hypoglycaemia, hypophosphatemia, metabolic acidosis, increase in uric acid, acute liver failure with breakdown of excretory and synthetic function, and death. Therefore, in patients with hereditary fructose intolerance the risk of treatment with Praxbind must be weighed against the potential benefit of such an emergency treatment.
Urinary protein testing. Praxbind causes transient proteinuria as a physiologic reaction to renal protein overflow after bolus/short-term application of 5 g idarucizumab intravenously (see Section 5.2 Pharmacokinetic Properties). The transient proteinuria is not indicative of renal damage, which should be taken into account for urine testing.
Re-elevation of coagulation parameters. In a limited number of patients, recurrence of plasma concentrations of unbound dabigatran and concomitant elevated coagulation parameters have occurred up to 24 hours after administration of idarucizumab (see Section 5.1 Pharmacodynamic Properties).
If reappearance of clinically relevant bleeding together with elevated coagulation parameters is observed after administration of 5 g Praxbind, administration of an additional 5 g dose of Praxbind may be considered. Similarly, patients who require a second emergency surgery/urgent procedure and have elevated coagulation parameters may receive an additional 5 g dose of Praxbind (see Section 4.2 Dose and Method of Administration).
Sodium. This medicinal product contains 2.2 mmol (or 50 mg) sodium per dose. This should be taken into consideration by patients on a controlled sodium diet.
Use in hepatic impairment. An impact of hepatic impairment, as determined by elevated liver function tests, on the pharmacokinetics of idarucizumab has not been observed. No dose adjustment is required in patients with hepatic impairment.
Idarucizumab has been studied in 58 patients with varying degrees of hepatic impairment. Compared to 272 patients without hepatic impairment, the median AUC of idarucizumab was changed by -6%, 37% and 10% in patients with AST/ALT elevations of 1 to < 2 x ULN (N = 34), 2 to < 3 x ULN (N = 3) and > 3 x ULN (N = 21), respectively. Based on pharmacokinetic data from 12 patients with liver disease, the AUC of idarucizumab was increased by 10% as compared to patients without liver disease.
Use in renal impairment. No dose adjustment is required in renally impaired patients. Renal impairment did not impact the reversal effect of idarucizumab.
In phase I studies Praxbind has been investigated in subjects with a creatinine clearance ranging from 44 to 213 mL/min. Subjects with a creatinine clearance below 44 mL/min have not been studied in phase I.
Depending on the degree of renal impairment the total clearance was reduced compared to healthy subjects, leading to an increased exposure of idarucizumab.
The method used to estimate renal function (CrCl in mL/min) during the clinical development of Praxbind was the Cockcroft-Gault method.
See Table 1.
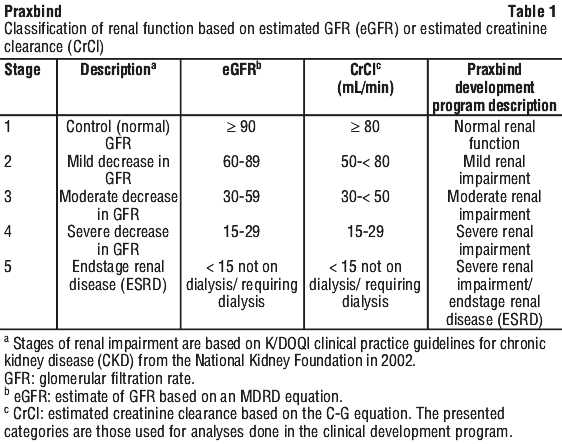
Based on these data and the extent of reversal of the anticoagulant effect of dabigatran in patients, renal impairment does not impact the reversal effect of idarucizumab.
Sex/race/bodyweight. Based on population pharmacokinetic analyses in healthy volunteers, sex, race and bodyweight do not have a clinically meaningful effect on the pharmacokinetics of idarucizumab.
Use in the elderly. Based on population pharmacokinetic analyses in healthy volunteers, age does not have a clinically meaningful effect on the pharmacokinetics of idarucizumab.
Paediatric use. The safety and efficacy of Praxbind in the paediatric population has not been established (see Section 5.1 Pharmacodynamic Properties).
Effects on laboratory tests. Idarucizumab showed no nonspecific binding to blood cells or to other thrombin substrates and did not exhibit thrombin like, prothrombotic effects in several in vitro assays. Coagulation test results (dTT, aPTT, ECT, thrombin time (TT), activated clotting time (ACT)) were comparable in the presence and absence of idarucizumab.
4.5 Interactions with Other Medicines and Other Forms of Interactions
No formal interaction studies with Praxbind and other medicinal products have been performed. Based on the pharmacokinetic properties and the high specificity in binding to dabigatran, clinically relevant interactions with other medicinal products are considered unlikely.
Preclinical investigations have shown no interactions with volume expanders, coagulation factor concentrates and anticoagulants other than dabigatran (see Section 5.1 Pharmacodynamic Properties).
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. Studies to assess the potential effects of idarucizumab on fertility have not been performed. Treatment related changes to reproductive tissues of either sex were not seen during repeat dose intravenous toxicity studies of up to four weeks in the rat and two weeks in monkeys. Additionally, no idarucizumab binding to human reproductive tissues was observed in a tissue cross reactivity study. Therefore, preclinical results do not suggest a risk to fertility or embryofetal development.
Use in pregnancy. (Category B2)
There are no data for the use of idarucizumab in pregnant women. Reproductive and developmental toxicity studies have not been performed, given the nature and the intended clinical use of the medicinal product. Idarucizumab may be used during pregnancy, if the expected clinical benefit outweighs the potential risks.
Use in lactation. It is unknown whether idarucizumab is excreted in human milk.
4.7 Effects on Ability to Drive and Use Machines
The effects of this medicine on a person's ability to drive and use machines were not assessed as part of its registration.
4.8 Adverse Effects (Undesirable Effects)
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
In phase III trials the safety of Praxbind has been evaluated in 504 patients (including one 17-year old patient), who had uncontrolled bleeding or required emergency surgery or procedures and were under treatment with dabigatran etexilate, as well as in 224 volunteers in phase I trials.
No adverse reactions have been identified.
Clinical trial experience. Three clinical trials in healthy volunteers have been completed, in which 224 subjects were treated with idarucizumab. In these trials during the treatment period the overall frequency of adverse events was similar between idarucizumab treated subjects (55/224, 25%) and placebo treated subjects (26/105, 25%).
Table 2 informs about adverse events reported in healthy volunteers treated with placebo alone, Praxbind alone and those treated either Praxbind alone or treated with Praxbind after pretreatment with dabigatran etexilate.
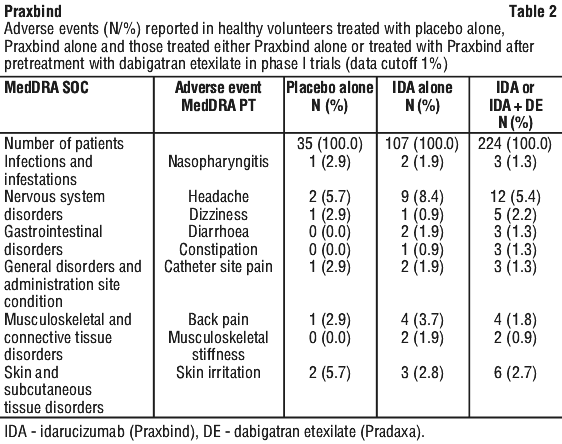
Thrombotic events. Thrombotic events were reported in 34 patients (23 out of the 34 patients were not on antithrombotic therapy at the time of the event) and in each of these cases, the thrombotic event could be attributed to the underlying medical condition of the patient.
Potential hypersensitivity. In the RE-VERSE AD trial, the following adverse events associated with hypersensitivity have been reported. A causal relationship to idarucizumab could not be established.
Mild symptoms of potential hypersensitivity: erythema 0.8%, pruritus 0.8%, respiratory distress 0.4%, mouth ulcerations 0.2%, respiratory arrest 0.2%, skin oedema 0.2%, wheezing 0.2%.
Moderate symptoms of potential hypersensitivity: bronchospasm 0.4%, generalised oedema 0.4%, localised oedema 0.4%, pruritus 0.4%, scrotal oedema 0.2%, urticaria 0.2%, wheezing 0.2%.
Severe symptoms of potential hypersensitivity: respiratory failure 1.2%, shock 0.4%, acute respiratory failure 0.2%, anaphylactic reaction 0.2%, anaphylactic shock 0.2%, circulatory collapse 0.2%, pneumonitis 0.2%.
Adverse events occurring in greater than 3% (total) of patients treated with dabigatran etexilate and experiencing uncontrolled bleeding (group A) or requiring emergency surgery or procedures (group B) are shown in Table 3.
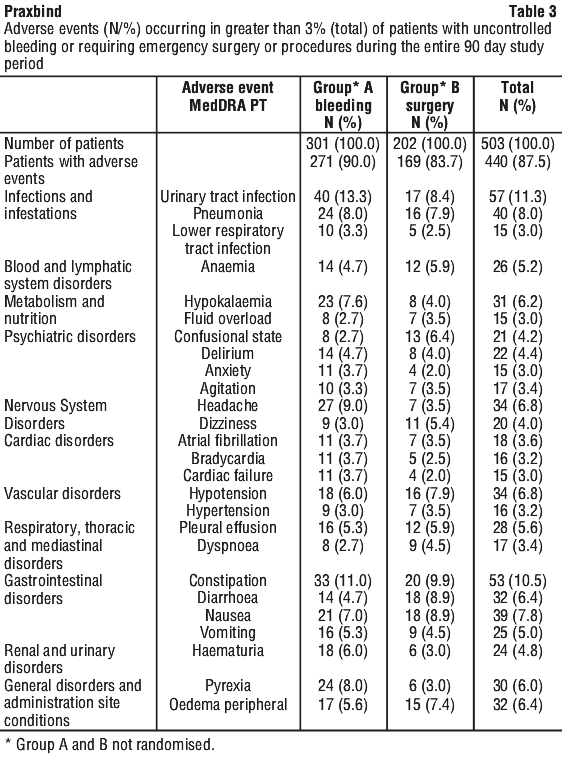
4.9 Overdose
For information on the management of overdose contact the Poisons Information Centre on 13 11 26 (Australia).
There is no clinical experience with overdoses of Praxbind.
The highest dose of Praxbind studied in healthy subjects was 8 g. No safety signals have been identified in this group.
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: all other therapeutic products, antidotes.
ATC code: V03AB37.
Mechanism of action. Idarucizumab is a specific reversal agent for dabigatran. It is a humanised monoclonal antibody fragment (Fab) that binds to dabigatran with very high affinity, approximately 300-fold higher than the binding affinity of dabigatran for thrombin at physiological pH (pH 7.4). The idarucizumab-dabigatran complex is characterised by a rapid on rate and extremely slow off rate resulting in a very stable complex. Idarucizumab specifically binds to dabigatran and its acylglucuronide metabolites and potently neutralises their anticoagulant effect.
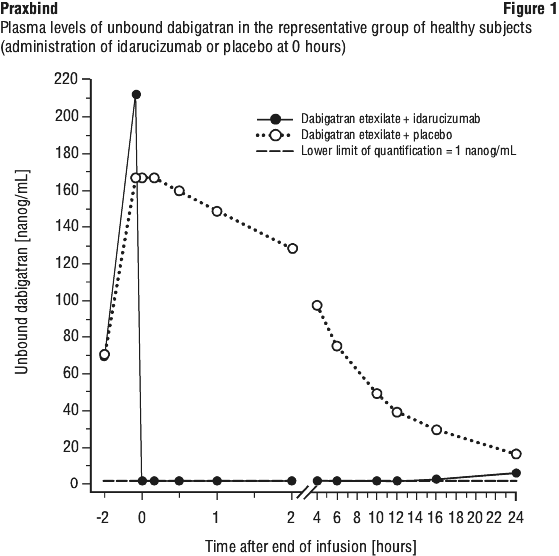
The pharmacodynamics of idarucizumab after administration of dabigatran etexilate were investigated in 141 subjects in phase I studies, of which data for a representative subgroup of 6 healthy subjects aged 45 to 64 years receiving a dose of 5 g via intravenous infusion are presented. The median peak dabigatran exposure in the investigated healthy subjects was in the range of a twice daily administration of 150 mg dabigatran etexilate in patients.
Effect of idarucizumab on the exposure and anticoagulant activity of dabigatran. Immediately after the administration of idarucizumab, the plasma concentrations of unbound dabigatran were reduced by more than 99%, resulting in levels with no anticoagulant activity.
The majority of the patients showed sustained reversal of dabigatran plasma concentrations up to 12 hours (≥ 90%). In a subset of patients, recurrence of plasma levels of unbound dabigatran and concomitant elevation of clotting tests was observed, possibly due to redistribution of dabigatran from the periphery. This occurred 1-24 hours after administration of idarucizumab mainly at timepoints ≥ 12 hours.
See Figure 1.
See Figure 2.
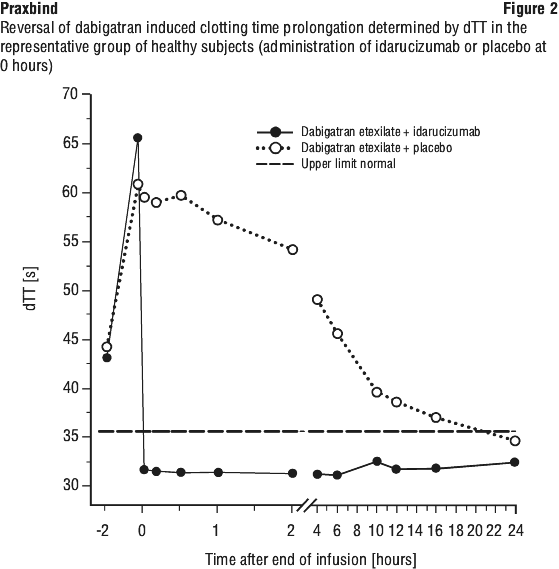
Readministration of dabigatran etexilate. 24 hours after the idarucizumab infusion, readministration of dabigatran etexilate resulted in expected anticoagulant activity.
Immunogenicity. Serum samples from 283 subjects in phase I trials (224 volunteers treated with idarucizumab) and 501 patients were tested for antibodies to idarucizumab before and after treatment.
Pre-existing antibodies with cross reactivity to idarucizumab were detected in approximately 12% (33/283) of the phase I subjects and 3.8% (19/501) of the patients. No impact on the pharmacokinetics or the reversal effect of idarucizumab or hypersensitivity reactions were observed.
Treatment emergent possibly persistent anti-idarucizumab antibodies with low titres were observed in 4% (10/224) of the phase I subjects and 1.6% (8/501) of the patients suggesting a low immunogenic potential of idarucizumab. In a subgroup of 6 phase I subjects, idarucizumab was administered a second time, two months after the first administration. No anti-idarucizumab antibodies were detected in these subjects prior to the second administration. In one subject, treatment emergent anti-idarucizumab antibodies were detected after the second administration. Nine patients were re-dosed with idarucizumab. All nine patients were re-dosed within 6 days after the first idarucizumab dose. None of the patients re-dosed with idarucizumab tested positive for anti-idarucizumab antibodies.
Preclinical pharmacodynamics. A trauma model in pigs was performed using a blunt liver injury after dosing with dabigatran to achieve supratherapeutic concentrations of about 10-fold of human plasma levels. Idarucizumab effectively and rapidly reversed the life threatening bleeding within 15 minutes after the injection. All pigs survived at idarucizumab doses of approximately 2.5 and 5 g. Without idarucizumab, the mortality in the anticoagulated group was 100%. When idarucizumab is present in less than equimolar concentrations, some residual dabigatran activity can reappear if haemostasis has not been achieved.
Preclinical investigations with idarucizumab have shown no interactions with:
colloid and crystalloid volume expanders (e.g. gelatin or hydroxyethyl starch);
coagulation factor concentrates, such as prothrombin complex concentrates (PCCs, e.g. 3 factor and 4 factor), activated PCCs (aPCCs) and recombinant factor VIIa;
other anticoagulants (e.g. thrombin inhibitors other than dabigatran, Factor Xa inhibitors including low molecular weight heparin, vitamin K antagonists, heparin).
Thus idarucizumab will not reverse the effects of other anticoagulants.
Clinical trials. Three randomised, double blind, placebo controlled phase I studies in 283 subjects (224 treated with idarucizumab) were conducted to assess the safety, efficacy, tolerability, pharmacokinetics and pharmacodynamics of idarucizumab, given alone or after administration of dabigatran etexilate. The investigated population consisted of healthy subjects and subjects exhibiting specific population characteristics covering age, bodyweight, race, sex and renal impairment. In these studies the doses of idarucizumab ranged from 20 mg to 8 g and the infusion times ranged from 5 minutes to 1 hour.
Representative values for pharmacokinetic and pharmacodynamic parameters were established on the basis of healthy subjects aged 45-64 years receiving 5 g idarucizumab (see Section 5.1 Pharmacodynamic Properties; Section 5.2 Pharmacokinetic Properties).
One prospective, open label, nonrandomised, uncontrolled study (RE-VERSE AD) was conducted to investigate the treatment of adult patients who presented with dabigatran related life threatening or uncontrolled bleeding (group A) or who required emergency surgery or urgent procedures (group B). The primary endpoint was the maximum percentage reversal of the anticoagulant effect of dabigatran within 4 hours after the administration of idarucizumab, based on central laboratory determination of diluted thrombin time (dTT) or ecarin clotting time (ECT). A key secondary endpoint was the restoration of haemostasis.
RE-VERSE AD included data for 503 patients: 301 patients with serious bleeding (group A) and 202 patients requiring an urgent procedure/surgery (group B). Approximately half of the patients were male. The median age was 78 years and the median creatinine clearance was 52.6 mL/min. 61.5% of patients in group A and 62.4% of patients in group B had been treated with dabigatran 110 mg twice daily.
Reversal was only evaluable for those patients showing prolonged coagulation times prior to idarucizumab treatment (ECT: N = 461 out of 503 patients; dTT N = 396 out of 503 patients; aPTT N = 373 out of 503 patients).
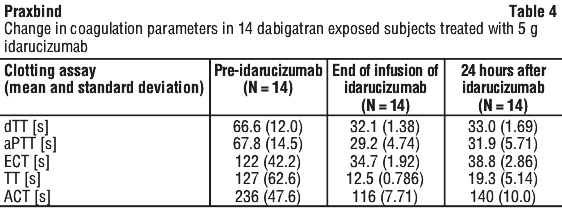
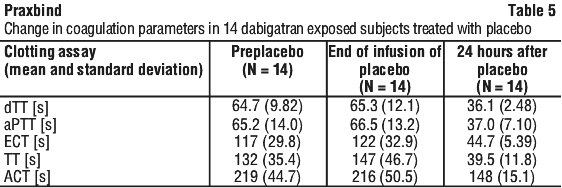
Most patients, in both groups A and B, achieved complete reversal of the anticoagulant effect of dabigatran (dTT: 98.7%; ECT: 82.2%; aPTT: 92.5% of evaluable patients, respectively) in the first 4 hours after administration of 5 g idarucizumab. Reversal effects were evident immediately after administration. In line with the re-elevation of unbound sum dabigatran concentrations (see Section 4.4 Special Warnings and Precautions for Use), re-elevation of coagulation parameters was observed in some patients. At 12 hours after idarucizumab administration, 41/457, 162/457 and 75/457 patients with available data had clotting times exceeding the upper limit of normal threshold for dTT, ECT and aPTT, respectively. At 24 hours, the numbers of patients were 79/449, 206/448 and 122/446 for dTT, ECT and aPTT, respectively. The clinical relevance of elevation of clotting tests without clinical symptoms of bleeding is difficult to interpret.
Figures 3, 4 and 5 show the reversal of dabigatran induced clotting time prolongation determined by dTT, ECT or aPTT in patients from the RE-VERSE AD study.
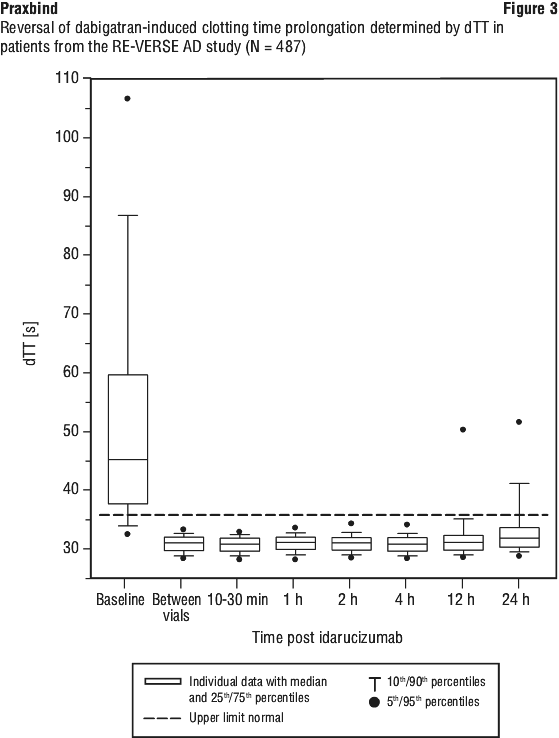
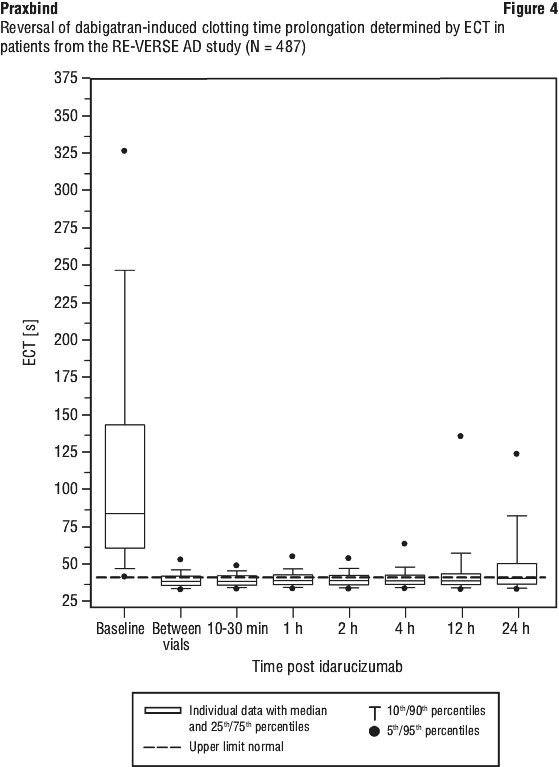
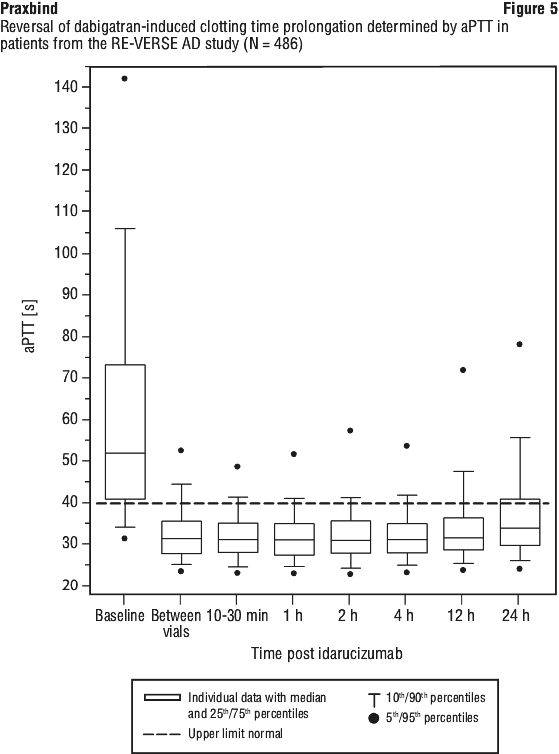
Of the total 503 patients, 101 patients died; each of these deaths could be attributed either as a complication of the index event or associated with comorbidities. Thrombotic events were reported in 34 patients (23 out of the 34 patients were not on antithrombotic therapy at the time of the event) and in each of these cases, the thrombotic event could be attributed to the underlying medical condition of the patient. Mild symptoms of potential hypersensitivity (pyrexia, bronchospasm, hyperventilation, rash or pruritus) were reported. A causal relationship to idarucizumab could not be established. All adverse events reported in greater than or equal to 3% of patients are summarised in Table 3 (see Section 4.8 Adverse Effects (Undesirable Effects)).
Paediatric population. One 17-year old patient (weight > 70 kg) was included in a single dose, open label, safety trial of intravenous administration of idarucizumab. The trial sought to enrol paediatric patients from clinical trials with dabigatran etexilate for the treatment and secondary prevention of venous thromboembolism (VTE). For inclusion, patients required rapid reversal of the anticoagulant effect of dabigatran. The 17-year-old female patient was treated with dabigatran etexilate for secondary prevention of VTE due to the presence of a clinical risk factor. A bleeding event required a surgical intervention and adequate haemostasis. Treatment with a weight-appropriate dose (5 g) of idarucizumab resulted in a rapid and complete reversal of the anticoagulant effect of dabigatran.
5.2 Pharmacokinetic Properties
The pharmacokinetics of idarucizumab were investigated in 224 subjects in phase I studies, of which data for a representative subgroup of 6 healthy subjects aged 45 to 64 years receiving a dose of 5 g via intravenous infusion are presented.
Distribution. Idarucizumab exhibited multiphasic disposition kinetics and limited extravascular distribution. Following the intravenous infusion of a 5 g dose, the geometric mean volume of distribution at steady-state (Vss) was 8.9 L (geometric coefficient of variation (gCV) 24.8%).
Metabolism. Several pathways have been described that may contribute to the metabolism of antibodies. All of these pathways involve biodegradation of the antibody to smaller molecules, i.e. small peptides or amino acids which are then reabsorbed and incorporated in the general protein synthesis.
Excretion. Idarucizumab was rapidly eliminated with a total clearance of 47.0 mL/min (gCV 18.4%), an initial half-life of 47 minutes (gCV 11.4%) and a terminal half-life of 10.3 hours (gCV 18.9%). After intravenous administration of 5 g idarucizumab, 32.1% (gCV 60.0%) of the dose was recovered in urine within a collection period of 6 hours and less than 1% in the following 18 hours. The remaining part of the dose is assumed to be eliminated via protein catabolism, mainly in the kidney.
After treatment with idarucizumab proteinuria has been observed. The transient proteinuria is a physiologic reaction to renal protein overflow after bolus/short-term application of 5 g idarucizumab intravenously. The transient proteinuria usually peaked about 4 hours after idarucizumab administration and normalised within 12-24 hours. In single cases the transient proteinuria persisted for more than 24 hours.
Special populations. Use in renal impairment. Total idarucizumab clearance was reduced in subjects with renal impairment compared to healthy subjects, leading to an increased exposure of idarucizumab. These findings were consistent with the available data from 347 patients in the RE-VERSE AD trial (see Section 4.4 Special Warnings and Precautions for Use, Use in renal impairment).
Elderly patients/sex/race/bodyweight. Based on population pharmacokinetic analyses in healthy volunteers, sex, age, race and bodyweight do not have a clinically meaningful effect on the pharmacokinetics of idarucizumab.
5.3 Preclinical Safety Data
Genotoxicity. Studies to evaluate the genotoxic potential of idarucizumab have not been performed. Based on its mechanism of action and the characteristics of proteins, no genotoxic effects are anticipated.
Carcinogenicity. The carcinogenic potential of idarucizumab has not been investigated in animal studies. Based on its mechanism of action and the characteristics of proteins, no carcinogenic effects are anticipated.
6 Pharmaceutical Particulars
6.1 List of Excipients
Praxbind also contains glacial acetic acid, polysorbate 20, sodium acetate trihydrate, sorbitol and water for injection.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store in a refrigerator at 2°C to 8°C. Do not freeze.
Store in the original package in order to protect from light.
6.5 Nature and Contents of Container
Praxbind 50 mg/mL solution for injection/infusion is presented as a nominal 50.0 mL fill volume in a 50 mL glass vial, closed with a coated rubber stopper and secured with an aluminium flip-off cap.
Praxbind is supplied in packs of 2 vials.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Idarucizumab is a humanised monoclonal antibody fragment (Fab) molecule derived from murine IgG1 isotype antibody molecule. Idarucizumab drug substance is a colourless to slightly yellow, clear to slightly opalescent solution. The final formulated idarucizumab drug substance has a pH of 5.5 and an osmolality of 270-330 mOsmol/kg. The melting point of the idarucizumab molecule is 84.4°C.
Chemical structure. Molecular formula: C2131H3299N555O671S11.
Molecular mass: 47,766 Da.
Structural formula: Light chain (amino acids 1-219) and heavy chain fragments (amino acids 1-225), covalently linked together by one disulfide bond between cysteine 225 of the heavy chain fragment and cysteine 219 of the light chain.

7 Medicine Schedule (Poisons Standard)
S4 - Prescription Only Medicine.
Date of First Approval
11 May 2016
Date of Revision
14 January 2022
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.