Qarziba
Brand Information
| Brand name | Qarziba |
| Active ingredient | Dinutuximab beta |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Qarziba.
Summary CMI
QARZIBA
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
▼ This medicine is new or being used differently. Please report side effects. See the full CMI for further details.
1. Why am I using QARZIBA?
QARZIBA contains the active ingredient dinutuximab beta. QARZIBA is used to treat neuroblastoma that has a high risk of coming back after a series of treatments and who have achieved at least a partial response.
For more information, see Section 1. Why am I using QARZIBA? in the full CMI.
2. What should I know before I use QARZIBA?
Do not use if you have ever had an allergic reaction to dinutuximab beta or any of the ingredients listed at the end of the CMI.
Do not take Qarziba if you have acute grade 3 or 4, or extensive long-lasting graft-versus-host disease.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use QARZIBA? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with QARZIBA and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use QARZIBA?
- The administration of the medicine will be started by a doctor or nurse while you are in hospital.
- QARZIBA will be given to you in five treatment courses of 35 days and the infusion will last 5 or 10 days in the beginning of each course. The recommended dose for patients weighing over 12 kg is 100 mg dinutuximab beta per square metre of body surface per treatment course. The recommended dose for patients weighing over 5 kg but below 12 kg is 3.3 mg/kg per course. The doctor will calculate your body surface area from your height and weight.
More instructions can be found in Section 4. How do I take QARZIBA? in the full CMI.
5. What should I know while using QARZIBA?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using QARZIBA? in the full CMI.
6. Are there any side effects?
Like all medicines, this medicine can cause side effects. You might notice the following when you first receive QARZIBA and during the course of treatment: pain, allergic reactions or other infusion-related reactions, leakage from small blood vessels (capillary leak syndrome), eye problems, problems with your nerves and spinal cord and brain problems (central nervous system, CNS).
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
▼ This medicine is subject to additional monitoring. This will allow quick identification of new safety information. You can help by reporting any side effects you may get. You can report side effects to your doctor, or directly at www.tga.gov.au/reporting-problems.
QARZIBA
Active ingredient(s): dinutuximab beta
Consumer Medicine Information (CMI)
This leaflet provides important information about using QARZIBA. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using QARZIBA.
Where to find information in this leaflet:
1. Why am I using QARZIBA?
2. What should I know before I use QARZIBA?
3. What if I am taking other medicines?
4. How do I use QARZIBA?
5. What should I know while using QARZIBA?
6. Are there any side effects?
7. Product details
1. Why am I using QARZIBA?
QARZIBA contains the active ingredient dinutuximab beta, which belongs to a group of medicines called monoclonal antibodies. These are proteins, which specifically recognise and bind to other unique proteins in the body. Dinutuximab beta binds to the molecule known as disialoganglioside 2 (GD2), which is present on cancer cells, and this activates the body's immune system, causing it to attack the cancer cells.
QARZIBA is used to treat neuroblastoma that has a high risk of coming back after a series of treatments and who have achieved at least a partial response.
Neuroblastoma is a type of cancer that grows from abnormal nerve cells in the body, in particular in the glands above the kidneys. It is one of the most common cancers in infancy.
Ask your doctor if you have any questions about why this medicine has been prescribed for you.
Your doctor may have prescribed it for another reason.
2. What should I know before I use QARZIBA?
Warnings
Do not use QARZIBA if:
- you are allergic to dinutuximab beta or any of the ingredients listed at the end of this leaflet.
- you have acute grade 3 or 4, or extensive long-lasting graft-versus-host disease. This disease is a reaction in which cells of transplanted tissue attack cells of the recipient.
- Always check the ingredients to make sure you can use this medicine.
Before receiving QARZIBA, you will have blood tests to check your liver, lung, renal and bone marrow functions.
Check with your doctor if you:
- have allergies to any other medicines, foods, preservatives or dyes.
- notice the following when you first receive QARZIBA and during the course of treatment:
Pain:
Pain is one of the most common side effects of QARZIBA. It usually occurs at the beginning of infusion. Therefore, your doctor will give you an appropriate pain treatment starting 3 days before and continuing during use of QARZIBA.
Allergic reactions or other infusion-related reactions:
Tell your doctor or nurse if you have any kind of reaction during or after the infusion, such as:
- fever, shivering and/or low blood pressure
- difficulties in breathing
- skin rash, hives
You will receive appropriate treatment to prevent these reactions and be closely monitored for these symptoms during infusion of QARZIBA.
Leakage from small blood vessels (capillary leak syndrome):
Leakage of blood components from small blood vessels may cause rapid swelling in arms, legs and other parts of the body. Rapid drop in blood pressure, light-headedness and breathing difficulties are further signs.
Eye problems:
You may notice changes to your vision.
Problems with your nerves:
You may notice numbness, tingling or burning in your hands, feet, legs or arms, reduced sensation or weakness with movement.
Spinal cord and brain problems (central nervous system, CNS):
Tell your doctor or nurse if you have any kind of CNS symptoms, such as: substantial prolonged neurological deficit without apparent reason such as muscle weakness or loss of muscle strength in the legs (or arms), or mobility problems or unusual sensations and numbness. Persistent or sudden onset of a headache, or progressive loss of memory and cognitive ability, subtle personality changes, inability to concentrate, lethargy, and progressive loss of consciousness
Symptoms of kidney failure:
Tell your doctor or nurse if you notice an altered frequency or absence of urination.
Tell your doctor immediately if you notice any of these problems.
Your doctor may decide to stop your treatment if you have any of the problems mentioned here. In some cases your treatment may be able to start again after a break or at a slower rate, but sometimes it may need to be stopped completely.
Your doctor will do blood tests and may do eye tests while you are taking this medicine.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
Talk to your doctor before you receive QARZIBA if you are of childbearing age. It is recommended to use contraception for 6 months after discontinuation of treatment with QARZIBA. You may only use QARZIBA if your doctor assesses that benefits outweigh risks for a foetus.
Tell your doctor if you are breastfeeding. Do not breastfeed during treatment with QARZIBA and for 6 months after the last dose. It is not known if the medicine can pass into breast-milk.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Do not use medicines that suppress the immune system from 2 weeks before the first dose of QARZIBA until 1 week after the last treatment course, unless prescribed by your doctor. Examples of medicines that suppress the immune system are corticosteroids used to reduce inflammation or prevent organ transplant rejection.
Avoid vaccinations during treatment with QARZIBA and for 10 weeks afterwards.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect QARZIBA.
4. How do I use QARZIBA?
How much QARZIBA is given
- A doctor experienced in the use of medicines to treat cancer will direct your treatment.
- QARZIBA will be given to you in five treatment courses of 35 days and the infusion will last 5 or 10 days in the beginning of each course. The recommended dose for patients weighing over 12 kg is 100 mg dinutuximab beta per square metre of body surface per treatment course. The recommended dose for patients weighing over 5 kg but below 12 kg is 3.3 mg/kg per course. The doctor will calculate your body surface area from your height and weight.
How QARZIBA is given
- The administration of the medicine will be started by a doctor or nurse while you are in hospital. It is given into one of your veins (intravenous infusion) usually by using special tubes (catheters) and a pump. During and after the infusion, you will be checked regularly for infusion-related side effects.
If you are given too much QARZIBA
As this medicine will be given to you by your doctor or nurse, it is unlikely that you will be given too much. If you think you or anybody else has been given too much QARZIBA, contact your doctor, pharmacist or the Poisons Information Centre who will advise you what to do.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using QARZIBA?
Things you should do
- Keep all of your doctor appointments so that your progress can be checked.
- Your doctor may do some tests from time to time to make sure the medicine is working and to prevent unwanted side effects.
- Remind any doctor, dentist or pharmacist you visit that you are using QARZIBA.
Things you should not do
- Do not have this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering.
If the medicine has expired or is damaged, your doctor will return it to the hospital pharmacist for disposal. - Do not use medicines that suppress the immune system from 2 weeks before the first dose of QARZIBA until 1 week after the last treatment course, unless prescribed by your doctor.
- Avoid vaccinations during treatment with QARZIBA and for 10 weeks afterwards.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how QARZIBA affects you.
QARZIBA has several side effects that may affect your ability to drive and use machines. Do not perform these activities if your ability to concentrate and react is affected.
Looking after your medicine
Store in a refrigerator (2°C - 8°C). Keep the vial in the outer carton in order to protect from light.
Once opened, QARZIBA is intended for immediate use. Opened vials not immediately used are to be discarded.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Keep it where young children cannot reach it.
Getting rid of any unwanted medicine
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Very common side effects | What to do |
Pain
| Speak to your doctor if you have any of these side effects and they worry you. |
Serious side effects
| Common side effects | What to do |
Allergic reactions
| Speak to your doctor if you have any of these side effects and they worry you. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What QARZIBA contains
| Active ingredient (main ingredient) | dinutuximab beta
|
| Other ingredients (inactive ingredients) |
|
Do not take this medicine if you are allergic to any of these ingredients.
What QARZIBA looks like
QARZIBA is a colourless to slightly yellow liquid provided in a clear glass vial with a rubber stopper and aluminium seal.
Each carton contains 1 single use vial. AUST R 321016.
Who distributes QARZIBA
QARZIBA is distributed/supplied in Australia by:
Recordati Rare Diseases Australia Pty Ltd
Level 10, 100 Arthur Street,
North Sydney, NSW, 2060, Australia
Phone: +61 (0) 408 061 403
rrdaustraliainfo@recordati.com
This leaflet was prepared in November 2025.
Brand Information
| Brand name | Qarziba |
| Active ingredient | Dinutuximab beta |
| Schedule | S4 |
▼ This medicinal product is subject to additional monitoring in Australia. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse events at www.tga.gov.au/reporting-problems.
MIMS Revision Date: 01 March 2026
1 Name of Medicine
Qarziba (dinutuximab beta).
2 Qualitative and Quantitative Composition
1 mL of concentrate contains 4.5 mg dinutuximab beta.
Each vial contains 20 mg dinutuximab beta in 4.5 mL.
Dinutuximab beta is a mouse-human chimeric monoclonal IgG1 antibody produced in a mammalian cell line (CHO) by recombinant DNA technology.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Concentrate for solution for infusion. Colourless to slightly yellow liquid.
4 Clinical Particulars
4.1 Therapeutic Indications
Qarziba is indicated for the treatment of high-risk neuroblastoma in patients who have previously received induction chemotherapy and achieved at least a partial response.
4.2 Dose and Method of Administration
Qarziba must be administered under the direction of a physician experienced in the use of oncological therapies. The infusion must be administered by a healthcare professional prepared to manage severe allergic reactions including anaphylaxis in an environment where full resuscitation services are immediately available.
Dosage. Treatment with Qarziba consists of 5 consecutive courses, each course comprising 35 days.
The individual dose is determined based on the body surface area and should be a total of 100 mg/m2 per course.
There are two possible administration schedules:
Continuous infusion: a single infusion given over the first 10 days of each course (a total of 240 hours) (10 mg/m2/day); or
Divided daily infusions: an 8-hour infusion once per day for the first 5 days of each course (20 mg/m2/day).
Administration by the continuous infusion method may reduce likelihood/severity of adverse effects.
Prior to starting each treatment course, the following clinical parameters should be evaluated and treatment should be delayed until these values are reached:
pulse oximetry > 94% on room air;
adequate bone marrow function: absolute neutrophil count ≥ 500/microL, platelet count ≥ 20,000/microL, haemoglobin > 8.0 g/dL;
adequate liver function: alanine aminotransferase (ALT)/ aspartate aminotransferase (AST) < 5 times upper limit of normal (ULN);
adequate renal function: creatinine clearance or glomerular filtration rate (GRF) > 60 mL/min/1.73 m2.
Method of administration. Qarziba is for intravenous infusion. The solution should be administered via a peripheral or central intravenous line. Other intravenously co-administered agents should be delivered via a separate infusion line (see Section 6.6 Special Precautions for Disposal).
For continuous infusions, the solution is administered at a rate of 2 mL per hour (48 mL per day) using an infusion pump.
For 8 hour daily infusions, the solution is administered at a rate of approximately 13 mL per hour.
Pre-medication should always be considered before starting each infusion (see Section 4.4 Special Warnings and Precautions for Use).
For instructions on dilution of the medicinal product before administration, see Section 6.4 Special Precautions for Storage.
Product is for single use in one patient only. Discard any residue.
Dosage adjustment. Based on the physician's evaluation of the severity of adverse drug reactions to dinutuximab beta, patients may undergo a dose reduction of 50% or a temporary interruption of the infusion. As a consequence, either the infusion period is prolonged or, if tolerated by the patient, the infusion rate may be increased up to 3 mL/h (continuous infusion), in order to administer the total dose. See Table 1.
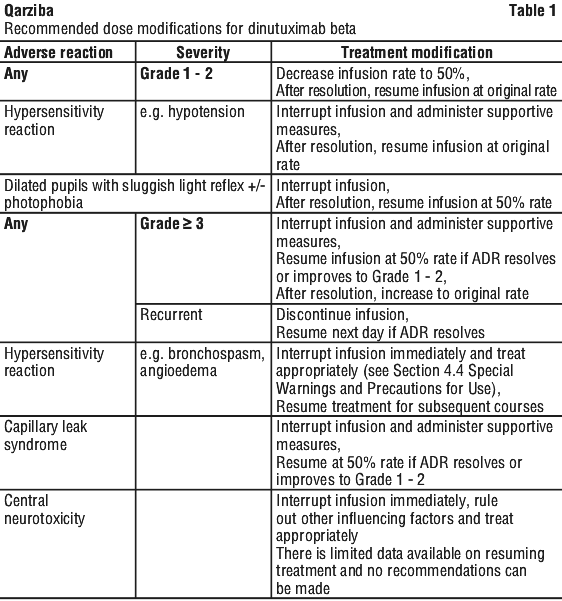
grade 3 or 4 anaphylaxis;
prolonged grade 2 peripheral motor neuropathy;
grade 3 peripheral neuropathy;
grade 3 vision eye toxicity;
grade 4 hyponatremia (< 120 mEq/L) despite appropriate fluid management, recurrent or grade 4 capillary leak syndrome (requires ventilator support);
severe central neurotoxicity that includes grade 3 or 4 with substantial prolonged neurological deficit without any detectable reason, recurrent grade 1-3 neurotoxicity, and permanent neurological deficit;
all grades of posterior reversible encephalopathy syndrome and transverse myelitis.
Renal and hepatic impairment. There are no data in patients with renal and hepatic impairment (see Section 5.2 Pharmacokinetic Properties).
Paediatric dose. Patients lighter than 12 kg received weight-based dosing in initial dinutuximab beta clinical studies: 0.33 mg/kg/day (continuously infused) for 10 days or 0.66 mg/kg/day (infused over 8 hours each day) for five days. BSA-based dosing is now recommended for all patients based on modelling and simulations, however, the model was developed using observed pharmacokinetic data that did not include any patients younger than 1 year or lighter than 9.1 kg. There is uncertainty regarding optimal dosing in such patients.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of Excipients.
Acute grade 3 or 4, or extensive chronic graft-versus-host disease (GvHD).
4.4 Special Warnings and Precautions for Use
Identified precautions. Pain. Neuropathic pain usually occurs at the beginning of the treatment and premedication with analgesics, including intravenous opioids, prior to each infusion of dinutuximab beta is required. A triple therapy, including nonopioid analgesics (according to WHO guidelines), gabapentin and opioids, is recommended for pain treatment. The individual dose may vary widely.
Nonopioid analgesics. Nonopioid analgesics should be used permanently during the treatment, e.g. paracetamol or ibuprofen.
Gabapentin. The patient should be primed with 10 mg/kg/day, starting 3 days prior to dinutuximab beta infusion. The daily dose of gabapentin is increased to 2 x 10 mg/kg/day orally, the next day and to 3 x 10 mg/kg/day orally, the day before the onset of dinutuximab beta infusion and thereafter. The maximum single dose of gabapentin is 300 mg. This dosing schedule should be maintained for as long as required by the patient.
Oral gabapentin should be tapered off after weaning off intravenous morphine infusion, at the latest after dinutuximab beta infusion therapy has stopped.
Opioids. Treatment with opioids is standard with dinutuximab beta. The first infusion day and course usually requires a higher dose than subsequent days and courses.
Before initiation of a continuous intravenous morphine infusion, a bolus infusion of 0.02 to 0.05 mg/kg/hour morphine should be started 2 hours before dinutuximab beta infusion.
Subsequently, a dosing rate of 0.03 mg/kg/hour is recommended concomitantly with dinutuximab beta infusion.
With daily infusions of dinutuximab beta, morphine infusion should be continued at a decreased rate (e.g. 0.01 mg/kg/h) for 4 hours after the end of dinutuximab beta infusion.
With continuous infusion, in response to the patient's pain perception, it may be possible to wean off morphine over 5 days by progressively decreasing its dosing rate (e.g. to 0.02 mg/kg/hour, 0.01 mg/kg/hour, 0.005 mg/kg/hour).
If continuous morphine infusion is required for more than 5 days, treatment should be gradually reduced by 20% per day after the last day of dinutuximab beta infusion.
After weaning off intravenous morphine, in case of severe neuropathic pain, oral morphine sulphate (0.2 to 0.4 mg/kg every 4 to 6 hours) can be administered on demand. For moderate neuropathic pain, oral tramadol may be administered.
Hypersensitivity reactions. Severe infusion-related reactions, including cytokine release syndrome (CRS), anaphylactic and hypersensitivity reactions, may occur despite the use of premedication. Occurrence of a severe infusion related reaction (including CRS) requires immediate discontinuation of dinutuximab beta therapy and may necessitate emergency treatment.
Cytokine release syndrome frequently manifests itself within minutes to hours of initiating the first infusion and is characterised by systemic symptoms such as fever, hypotension and urticaria.
Anaphylactic reactions may occur as early as within a few minutes of the first infusion with dinutuximab beta and are commonly associated with bronchospasm and urticaria.
Premedication. Antihistamine premedication (e.g. diphenhydramine) should be administered by intravenous injection approximately 20 minutes before starting each dinutuximab beta infusion. It is recommended that antihistamine administration be repeated every 4 to 6 hours as required during dinutuximab infusion.
Patients should be closely monitored for anaphylaxis and allergic reactions, particularly during the first and second treatment course.
Treatment of hypersensitivity reactions. Intravenous antihistamine, epinephrine (adrenaline) and prednisolone for intravenous administration should be immediately available at the bedside during administration of dinutuximab beta to manage life-threatening allergic reactions. It is recommended that treatment for such reactions include prednisolone administered by intravenous bolus, and epinephrine administered by intravenous bolus every 3 to 5 minutes as necessary, according to clinical response. In case of bronchial and/or pulmonary hypersensitivity reaction, inhalation with epinephrine (adrenaline) is recommended and should be repeated every 2 hours, according to clinical response.
Capillary leak syndrome (CLS). CLS is characterised by a loss of vascular tone and extravasation of plasma proteins and fluid into the extravascular space. CLS usually develops within hours after initiation of treatment, while clinical symptoms (i.e. hypotension, tachycardia) are reported to occur after 2 to 12 hours. Careful monitoring of circulatory and respiratory function is required.
Neurological disorders of the eye. Eye disorders may occur as dinutuximab beta binds to optic nerve cells. No dose modification is necessary in the case of an impaired visual accommodation that is correctable with eye glasses, as long as this is judged to be tolerable.
Treatment must be interrupted in patients who experience Grade 3 vision toxicity (i.e. subtotal vision loss per toxicity scale). In case of any eye problems, patients should be referred promptly to an ophthalmology specialist.
Peripheral neuropathy. Occasional occurrences of peripheral neuropathy have been reported with Qarziba. Cases of motor or sensory neuropathy lasting more than 4 days must be evaluated and noninflammatory causes, such as disease progression, infections, metabolic syndromes and concomitant medication, should be excluded.
Treatment should be permanently discontinued in patients experiencing any objective prolonged weakness attributable to dinutuximab beta administration. For patients with moderate (Grade 2) neuropathy (motor with or without sensory), treatment should be interrupted and may be resumed after neurologic symptoms resolve.
Central neurotoxicity. Central neurotoxicity has been reported following treatment with Qarziba. If central neurotoxicity occurs the infusion should be interrupted immediately and the patient treated symptomatically, other influencing factors such as active infection, metastatic spread of neuroblastoma to CNS, neurotoxic concomitant medications should be ruled out.
Treatment with dinutuximab beta should be permanently discontinued following the occurrence of severe neurotoxicity that includes grade 3 or 4 central neurotoxicity with substantial prolonged neurological deficit without any detectable reason, recurrent grade 1-3 neurotoxicity and/or permanent neurological deficit and all grades of posterior reversible encephalopathy syndrome and transverse myelitis.
Systemic infections. Patients are likely to be immunocompromised as a result of prior therapies. As they typically have a central venous catheter in situ, they are at risk of developing systemic infection.
Patients should have no evidence of systemic infection and any identified infection should be under control before starting therapy.
Haematologic toxicities. Occurrence of haematologic toxicities has been reported with Qarziba, such as erythropenia, thrombocytopenia or neutropenia. Grade 4 haematologic toxicities, improving to at least Grade 2 or baseline values by start of next treatment course, do not require dose modification.
Laboratory abnormalities. Regular monitoring of liver function and electrolytes is recommended.
Atypical haemolytic uraemic syndrome. Atypical haemolytic uraemic syndrome (aHUS) has been reported in patients who received dinutuximab beta, in some cases with fatal outcome. Signs and symptoms of aHUS should be monitored for. If aHUS is diagnosed, prompt treatment is required and dinutuximab beta should be permanently discontinued.
Use in the elderly. No data available.
Paediatric use. Data in children aged less than 12 months are limited.
Effects on laboratory tests. No data available.
4.5 Interactions with Other Medicines and Other Forms of Interactions
No interaction studies have been performed. A risk for indirect reduction of CYP activity due to higher TNF-α and IL-6 levels and, therefore, interactions with concomitantly used medicinal products, cannot be excluded.
Corticosteroids. Due to their immunosuppressive activity, concomitant treatment with corticosteroids is not recommended within 2 weeks prior to the first treatment course until 1 week after the last treatment course with dinutuximab beta, except for life-threatening conditions.
Vaccinations. Vaccinations should be avoided during administration of dinutuximab beta until 10 weeks after the last treatment course, due to immune stimulation through dinutuximab beta and possible risk for rare neurological toxicities.
Intravenous immunoglobulin. Concomitant use of intravenous immunoglobulins is not recommended as they may interfere with dinutuximab beta-dependent cellular cytotoxicity.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. The effects of dinutuximab beta on fertility in humans are unknown. In animals, dedicated fertility studies have not been conducted, but no adverse effects on reproductive organs were observed in toxicity studies performed in Guinea pig and cynomolgus monkey.
Qarziba should not be used in women of childbearing potential not using contraception. It is recommended that women of childbearing potential use contraception for 6 months after discontinuation of treatment with dinutuximab beta.
Use in pregnancy. (Category C)
There are no data on pregnant women. No animal data are available on teratogenicity or embryotoxicity. Dinutuximab beta target (GD2) is expressed on neuronal tissues, especially during embryofetal development, and may cross the placenta; therefore, Qarziba may cause fetal harm when administered to pregnant women.
Qarziba should not be used during pregnancy.
Use in lactation. There are no data on lactating women. It is unknown whether dinutuximab beta is excreted in human milk. Breast-feeding should be discontinued during treatment with Qarziba and for 6 months after the last dose.
4.7 Effects on Ability to Drive and Use Machines
Dinutuximab beta has major influence on the ability to drive and use machines. Patients should not use or drive machines during treatment with dinutuximab beta.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile. The safety of dinutuximab beta has been evaluated in 791 patients with high-risk and relapsed/refractory neuroblastoma, who received it as a continuous infusion (212) or as repeated daily infusions (416). It was combined with 13-cis retinoic in most patients and with IL-2 in 307 patients.
The most common adverse reactions were pyrexia (86%) and pain (57%) that occurred despite analgesic treatment. Other frequent adverse reactions were hypersensitivity (74.1%), vomiting (55%), diarrhoea (52%), capillary leak syndrome (36%), anaemia (49%), neutropenia (46%), thrombocytopenia (42%) and hypotension (41%).
Tabulated list of adverse reactions. Adverse reactions reported in clinical trials and post-marketing are listed by system organ class and by frequency and summarised in Table 2. These adverse reactions are presented by MedDRA system organ class and frequency. Frequency categories are defined as: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100) and not known (cannot be estimated from the available data). Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness.
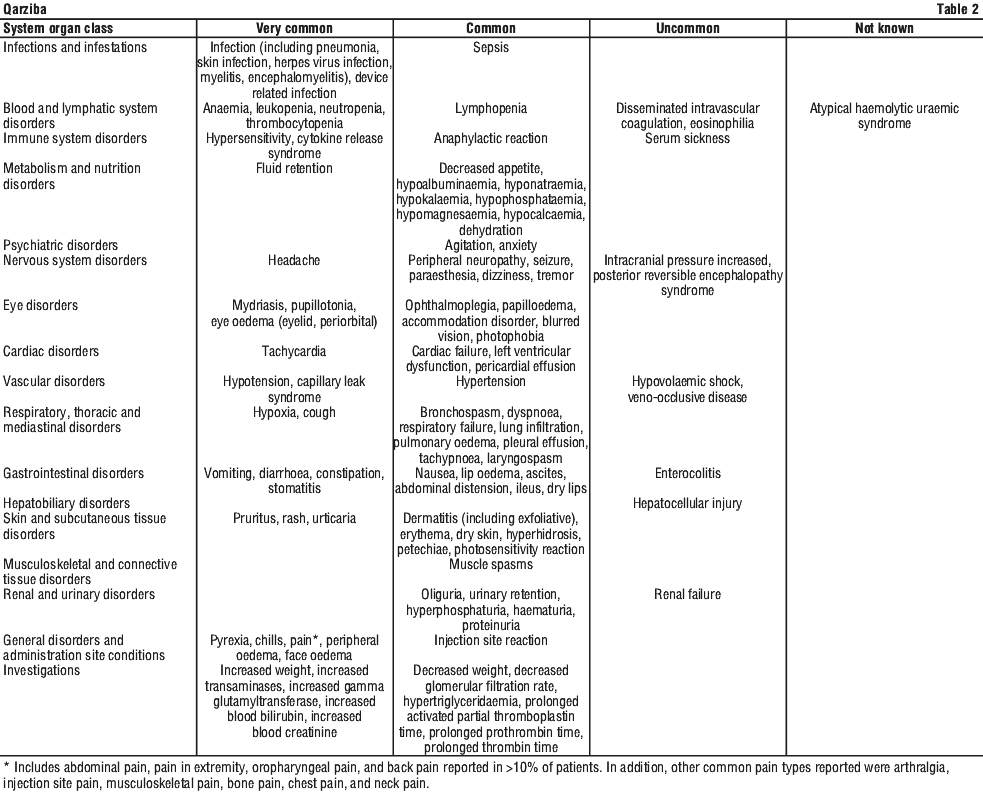
Pain. Pain typically occurs during the first infusion of dinutuximab beta and decreases over the treatment courses. Most commonly, patients reported abdominal pain, pain in the extremities, back pain, chest pain, or arthralgia.
Capillary leak syndrome (CLS). Overall, 10% of CLS were severe (grade 3-4) and their frequency decreased over the treatment courses.
Eye problems. These included impaired visual accommodation that is correctable with eyeglasses, as well as mydriasis (2%), periorbital oedema and eyelid oedema (3%), blurred vision (3%) or photophobia (3%), which were usually reversible after treatment discontinuation. Severe eye disorders were also reported including ophthalmoplegia (2%) and optic atrophy.
Peripheral neuropathy. Both motor and sensory peripheral neuropathies have been reported, overall in 9% of the patients. Most events were of grade 1-2 and resolved.
Central neurotoxicity. Reports of central neurotoxicity and severe neurotoxicity have been received including posterior reversible encephalopathy syndrome (0.7%) and seizures (1.7%).
Safety profile with and without IL-2. The combination of Qarziba with IL-2 increases the risk of adverse drug reactions compared to Qarziba without IL-2, especially for pyrexia (94% vs. 80%), CLS (45% vs. 20%), pain related to dinutuximab beta (70% vs. 62%), hypotension (44% vs. 27%), and peripheral neuropathy (9% vs. 5%), respectively.
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
4.9 Overdose
For information on the management of overdose, contact the Poisons Information Centre on 131126 (Australia) or 0800 764 766 (New Zealand).
No cases of dinutuximab beta overdose have been reported.
In the case of overdose, patients should be carefully observed for signs or symptoms of adverse reactions and supportive care administered, as appropriate.
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Antineoplastic agents, monoclonal antibodies, ATC code: L01FX06.
Mechanism of action. Dinutuximab beta is a chimeric monoclonal IgG1 antibody that is specifically directed against the carbohydrate moiety of disialoganglioside 2 (GD2), which is overexpressed on neuroblastoma cells.
Pharmacodynamic effects. Dinutuximab beta has been shown in vitro to bind to neuroblastoma cell lines known to express GD2 and to induce both complement dependent cytotoxicity (CDC) and antibody dependent cell-mediated cytotoxicity (ADCC). In the presence of human effector cells, including peripheral blood mononuclear cells from normal human donors, dinutuximab beta was found to mediate the lysis of human neuroblastoma and melanoma cell lines expressing GD2 in a dose-dependent manner. Additionally, in vivo studies demonstrated that dinutuximab beta could suppress liver metastasis in a syngeneic liver metastasis mouse model.
Neurotoxicity associated to dinutuximab beta is likely due to the induction of mechanical allodynia that may be mediated by the reactivity of dinutuximab beta with the GD2 antigen located on the surface of peripheral nerve fibres and myelin.
Clinical trials. The efficacy of dinutuximab beta has been evaluated in a randomised controlled trial comparing the administration of dinutuximab beta with or without IL-2 in the first-line treatment of patients with high-risk neuroblastoma and in two single-arm studies in the relapsed/refractory setting.
First-line patients who received autologous stem cell transplantation. In study 3, patients with high-risk neuroblastoma were enrolled after they had received induction chemotherapy and achieved at least a partial response, then myeloablative therapy and stem cell transplantation. Patients with progressive disease were excluded. Dinutuximab beta was administered at a dose of 20 mg/m2/day on 5 consecutive days, given by 8-hour intravenous infusion in a 5-week treatment course, and was combined with 13-cis-RA and with or without additional subcutaneous IL-2 at the same posologies as in the previous studies.
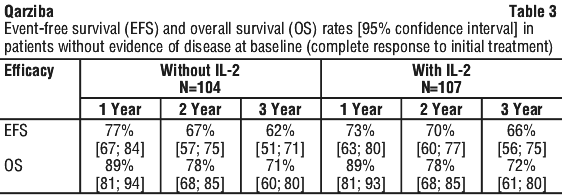
A total of 370 patients were randomised and received treatment. These included 64% male and 36% female patients with a median age of 3 years (0.6 to 20); 89% had a tumour INSS stage 4 and MYCN amplification was reported in 44% of the cases. The primary efficacy endpoint was 3-year EFS and secondary endpoint was OS. EFS and OS rates are presented in Tables 3 and 4 according to the evidence of disease at baseline.
For patients without evidence of disease at baseline, addition of IL-2 did not improve EFS and OS.

No clear associations between ADA response and relevant Selected Safety Events were observed.
From an efficacy and safety perspective, there is no rationale for adjusting or stopping treatment on the basis of measured ADA responses.
5.2 Pharmacokinetic Properties
Dinutuximab beta has been investigated using short-term infusions (STI - five days of eight hour infusions at 20 mg/m2/day) and long-term infusions (LTI - ten days of continuous infusion at 100 mg/m2).
Absorption. Dinutuximab beta is administered as an intravenous infusion. The maximum concentration (mean (± SD)) at the end of the long-term infusion was 11.2 (± 3.3) mg/L. Other routes of administration have not been investigated.
Distribution. The population mean (± SD) estimate for the central volume of distribution was 2.04 (± 1.05) L and for the peripheral volume of distribution 2.65 (± 1.01) L.
Biotransformation. The metabolism of dinutuximab beta has not been investigated. As a protein, dinutuximab beta is expected to be metabolised to small peptides and individual amino acids by ubiquitous proteolytic enzymes.
Elimination. The clearance after the LTI was 0.72 (± 0.24) L/d/m2. The accumulation ratio for Cmax was 1.13 (± 0.54) after 5 LTI courses (mean (± SD)). The apparent terminal elimination t1/2 was 8.7 (± 2.6) days (mean (± SD)). The clearance of dinutuximab beta increased in the presence of high anti-drug antibody titres regardless of neutralising activity. (See Section 5.1, Immunogenicity.)
Linearity/non-linearity. In one study, variations in dose of the first infusion revealed a dose-proportional increase in exposure (AUC∞) up to the recommended dose of 100 mg/m2 per course for 10 days.
Specific populations. The age of patients ranged from 1 to 27 years (median 6 years). Body weight ranged from 9 to 75 kg (median 18.5 kg) and body-surface area ranged from 0.44 to 1.94 m2 (median 0.75 m2). A two-compartment population PK model with first-order elimination from the central compartment was developed using the data from 224 patients in four studies (STI 30 patients, LTI 194 patients). Volume and clearance parameters increased across the ranges with increasing body size. Body weight and ADA titre were covariates for clearance while body weight, age and IL-2 co-administration were covariates for volume of distribution.
Age. The population pharmacokinetic analyses showed comparable exposure to dinutuximab beta in patients of all ages studied when dosed at 100 mg/m2.
Gender. The population pharmacokinetic analysis with 89 female (40%) and 135 male (60%) patients showed no clinically meaningful effect of gender on dinutuximab beta pharmacokinetics.
Race. Since the PK analysis population was predominantly Caucasian (92.9%) race was not formally examined as a potential PK covariate.
Weight. Dosing on the basis of body surface area provides consistent exposure across studied populations.
Renal impairment. No formal studies have been conducted in patients with renal impairment. Renal function was not a significant covariate in population pharmacokinetic analyses that included patients with normal renal function and mild renal impairment.
Hepatic impairment. No formal studies have been conducted in patients with hepatic impairment. Subjects with ALT > 3 x ULN had comparable pharmacokinetics as subjects with ALT ≤ 3 x ULN.
5.3 Preclinical Safety Data
General toxicology. Dinutuximab beta has been administered to male and female juvenile Guinea pigs, as well as male and female young cynomolgus monkeys, as repeat-dose regimens that exceeded the recommended clinical dose. Findings of note included changes (decrease) in thymus weight as well as bone marrow changes (atrophy affecting myeloid and erythroid precursor cell lines), reduced activity, food consumption and body weight gain possibly due to pain associated with drug treatment. The bone marrow changes were slight to severe and recovered after cessation of dosing. No effects on cardiovascular functions (ECG, blood pressure) were observed in monkeys.
Genotoxicity. No mutagenicity studies in animals have been performed with dinutuximab beta.
Carcinogenicity. No carcinogenicity studies in animals have been performed with dinutuximab beta.
6 Pharmaceutical Particulars
6.1 List of Excipients
Histidine, sucrose, polysorbate 20, water for injections, hydrochloric acid (for pH adjustment).
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those mentioned in Section 6.4 Special Precautions for Storage.
6.3 Shelf Life
Unopened vial. 4 years.
Diluted solution (solution for infusion). Chemical and physical in-use stability has been demonstrated for up to 48 hours at 25°C (50 mL syringe) and for up to 7 days at 37°C (250 mL infusion bag), after cumulative storage in a refrigerator (2°C to 8°C) for 72 hours (see Section 6.4 Special Precautions for Storage).
To reduce microbiological hazard, use as soon as practicable after preparation. If storage is necessary, hold at 2-8°C for not more than 24 hours.
6.4 Special Precautions for Storage
Store at 2°C to 8°C (Refrigerate. Do not freeze).
Keep the vial in the outer carton in order to protect from light.
For storage conditions after dilution of the medicinal product, see Section 6.3 Shelf Life.
Opened vials not immediately used are to be discarded.
Special precautions for handling. The solution for infusion must be prepared under aseptic conditions. The solution must not be exposed to direct sunlight or heat.
The patient specific daily dose of Qarziba is calculated based on body surface area (see Section 4.2 Dose and Method of Administration).
Qarziba should be diluted aseptically to the patient specific concentration/dose with sodium chloride 9 mg/mL (0.9%) solution for infusion containing 1% human albumin (e.g. 5 mL of human albumin 20% per 100 mL sodium chloride solution).
For continuous infusions, the solution for infusion can be prepared freshly on a daily basis, or sufficient for up to 5 days of continuous infusion. An in-line filter of 0.2 micrometre must be used in combination with infusion bags throughout the duration of the 5 days administration (or shorter, if applicable). The daily dose is 10 mg/m2. The amount of solution to be infused per day (within a treatment course of 10 consecutive days) should be 48 mL; with 240 mL for a 5-day dose. It is recommended to prepare 50 mL solution in a 50 mL syringe, or 250 mL in an infusion bag suitable for the employed infusion pump, i.e. an overfill of 2 mL (syringe) or 10 mL (infusion bag) to allow for dead volumes of the infusion systems.
For repeated daily 8-hour infusions, the daily dose is 20 mg/m2 and the calculated dose should be diluted in 100 mL sodium chloride 9 mg/mL (0.9%) containing 1% human albumin.
The solution for infusion should be administered via a peripheral or central intravenous line.
Other intravenously co-administered agents should be delivered via a separate infusion line. The container should be inspected visually for particulates prior to administration. It is recommended that a 0.22 micrometre in-line filter is used during infusion.
For continuous infusions, any medical device suitable for infusion at a rate of 2 mL per hour can be used, e.g. syringe infusion pumps/infusors, electronic ambulatory infusion pumps. Note that elastomeric pumps are not considered suitable in combination with in-line filters.
6.5 Nature and Contents of Container
Clear Type I glass vial (6 mL) with a halobutyl rubber stopper and aluminium flip-off cap, containing a minimum extractable volume of 4.5 mL concentrate for solution for infusion.
Each carton contains 1 single use vial.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
Chemical structure.
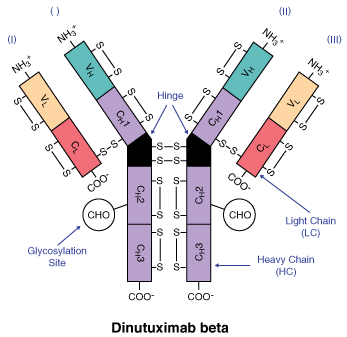
Molecular formula: C6422H9978N1722O2006S48 (without glycosylation).
C6546H10182N1730O2094S48 (including glycosylation).
7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.
Date of First Approval
02 April 2020
Date of Revision
27 January 2026
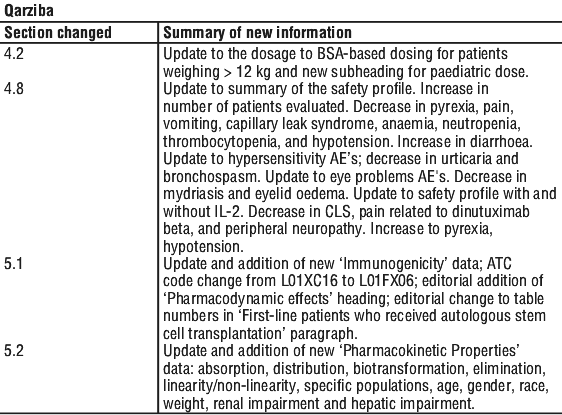
Summary Table of Changes
Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.