Steqeyma
Brand Information
| Brand name | Steqeyma |
| Active ingredient | Ustekinumab |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Steqeyma.
Summary CMI
STEQEYMA®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I using STEQEYMA?
STEQEYMA contains the active ingredient ustekinumab. STEQEYMA is used in adults and paediatric patients (children and adolescents) 6 years and older with moderate to severe plaque psoriasis that is chronic (doesn't go away), adults with active psoriatic arthritis (an inflammatory disease of the joints that is usually accompanied by psoriasis), moderately to severely active Crohn's disease (an inflammatory disease of the bowel) and moderate to severe ulcerative colitis (an inflammatory disease of the bowel).
For more information, see Section 1. Why am I using STEQEYMA? in the full CMI.
2. What should I know before I use STEQEYMA?
Do not use if you have ever had an allergic reaction to ustekinumab or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use STEQEYMA? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with STEQEYMA and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use STEQEYMA?
- STEQEYMA is injected under the skin (subcutaneous) More instructions can be found in Section 4. How do I use STEQEYMA? in the full CMI.
5. What should I know while using STEQEYMA?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Drinking alcohol |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using STEQEYMA? in the full CMI.
6. Are there any side effects?
Side effects that require urgent medical attention include: Signs of an allergic reaction, swollen face, lips, mouth or throat, cough, shortness of breath and fever.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
STEQEYMA® (Ste-QEY-ma)
Active ingredient: ustekinumab (rch)
Consumer Medicine Information (CMI)
This leaflet provides important information about using STEQEYMA. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using STEQEYMA.
Where to find information in this leaflet:
1. Why am I using STEQEYMA?
2. What should I know before I use STEQEYMA?
3. What if I am taking other medicines?
4. How do I use STEQEYMA?
5. What should I know while using STEQEYMA?
6. Are there any side effects?
7. Product details
1. Why am I using STEQEYMA?
STEQEYMA contains the active ingredient ustekinumab. Ustekinumab is a monoclonal antibody. Monoclonal antibodies are proteins that recognise and bind to other unique proteins.
Ustekinumab blocks the action of two proteins in your body called interleukin 12 (IL-12) and interleukin 23 (IL-23). IL-12 and IL-23 are made by your body's immune system. In people with psoriasis, psoriatic arthritis, and Crohn's disease or ulcerative colitis, IL-12 and IL-23 can cause their immune system to attack normal healthy parts of their body. Ustekinumab can block IL-12 and IL-23 from causing the immune system to attack.
STEQEYMA is used in adults and paediatric patients (children and adolescents) 6 years and older with moderate to severe plaque psoriasis that is chronic (doesn't go away), adults with active psoriatic arthritis (an inflammatory disease of the joints that is usually accompanied by psoriasis), moderately to severely active Crohn's disease (an inflammatory disease of the bowel) and moderate to severe ulcerative colitis (an inflammatory disease of the bowel).
2. What should I know before I use STEQEYMA?
Warnings
Do not use STEQEYMA if:
- you are allergic to ustekinumab, or any of the ingredients listed at the end of this leaflet.
- Always check the ingredients to make sure you can use this medicine.
- you have an infection.
Check with your doctor if you:
- have any kind of infection, even if it is very minor
- have an infection that won't go away or a history of infection that keeps coming back
- have had TB (tuberculosis), or if you have recently been near anyone who might have TB
- have or have had any type of cancer
- have any new or changing lesions within psoriasis areas or on normal skin
- recently received or are scheduled to receive a vaccine. Patients receiving STEQEYMA should not receive live vaccines. Tell your doctor if anyone in your house needs a vaccine. The viruses in some vaccines can spread to people with a weakened immune system and can cause serious problems
- are receiving allergy immunotherapy
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant. Ask your doctor for advice before taking this medicine.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
STEQEYMA may be used during a pregnancy if needed. STEQEYMA may pass into your breastmilk in very small amounts. Women who are breastfeeding should talk to their doctor about whether or not to use STEQEYMA as it might be excreted in breast milk.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect STEQEYMA.
You should not receive a live vaccine while taking STEQEYMA. If you used STEQEYMA while pregnant, tell you baby's doctor about your STEQEYMA use before the baby receives any vaccines, including live vaccines, such as the BCG vaccines (used to prevent tuberculosis), rotavirus vaccine or any other live vaccines.
4. How do I use STEQEYMA?
How much to take / use
- For the treatment of psoriasis or psoriatic arthritis STEQEYMA is given by injection just under the skin (subcutaneously).
- For the treatment of Crohn's Disease and ulcerative colitis, the first dose of STEQEYMA will be given by an intravenous infusion, which means that the medicine will be given to you through a needle placed in a vein. After this starting dose, all future doses of STEQEYMA are given by injection just under the skin (subcutaneously).
- All STEQEYMA injections are for single use in one patient only.
- STEQEYMA is intended for use under the guidance and supervision of your doctor. In children and adolescents 6 years and older with psoriasis, it is recommended that STEQEYMA be administered by a health care provider.
- When you start treatment, STEQEYMA may be injected by your healthcare provider. If your doctor determines that it is appropriate, you or your caregiver may be able to administer it under the skin yourself if you wish, after proper training in injection technique. (See How much to use and Injecting STEQEYMA under the skin yourself).
- It is important to remember that the first dose of STEQEYMA for the treatment of Crohn's Disease or ulcerative colitis (intravenous infusion) will always be administered by your healthcare provider.
How much to use
- Your doctor will determine the correct dose of STEQEYMA for you and how often you should receive it. Make sure to discuss with your doctor when you will receive injections and to come in for all your scheduled follow-up appointments.
For children and adolescents aged 6 years or older with psoriasis:
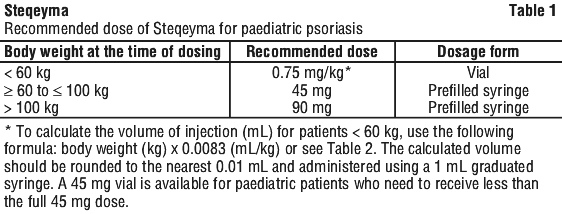
- The doctor will work out the right dose for you, including the amount (volume) of STEQEYMA to be injected to give the right dose. The right dose for you will depend on your body weight at the time each dose is given. If you weigh less than 60 kg, the recommended dose is 0.75 mg of STELARA per kg body weight.
- If you weigh 60 kg to 100 kg, the recommended dose is 45 mg STEQEYMA.
- If you weigh more than 100 kg, the recommended dose is 90 mg STEQEYMA.
- After the starting dose, you will have the next dose 4 weeks later, and then every 12 weeks.
- Follow the instructions provided and use STEQEYMA until your doctor tells you to stop.
How to use STEQEYMA
- STEQEYMA is injected under the skin (subcutaneous injection).
- It can be injected by the patient, or by someone else, such as a family member, friend or carer.
- Your first STEQEYMA injection may be administered by your healthcare provider. In children and adolescents 6 years and older, it is recommended that all doses of STEQEYMA be administered by a healthcare provider. However, your healthcare provider may decide that it is right for you or your caregiver to learn how to inject STEQEYMA under the skin (subcutaneously) yourself. If you would like to self-inject STEQEYMA under your skin, you or your caregiver must be trained by a healthcare professional to prepare an injection and give it to yourself. If you have not been trained, please contact your healthcare provider to schedule a training session. Call your healthcare provider if you have any questions about giving yourself an injection.
- STEQEYMA is given alone and not mixed with other liquids for injection.
STEQEYMA should not be used:
- After the expiry date on the label,
- If the seal is broken,
- If the liquid is discoloured, cloudy or you can see other particulate matter floating in it,
- If you know, or think that it might have been exposed to extreme temperatures (such as being accidentally frozen or heated).
Do not shake STEQEYMA at any time.
- Prolonged vigorous shaking may damage the product.
If STEQEYMA has been shaken vigorously, don't use it. STEQEYMA is not to be mixed with other liquids for injection. - STEQEYMA injection under the skin (subcutaneous) is available in a vial, pre-filled syringe and pre-filled pen.
- 'Instructions for Use' for the vial, pre-filled syringe and pre-filled pen are provided below.
- STEQEYMA pre-filled pen is not intended for use in children below 6 years of age.
When to use STEQEYMA
- Follow the instructions provided and use STEQEYMA until your doctor tells you to stop.
STEQEYMA is available in a vial for intravenous infusion which will be administered by your healthcare provider.
STEQEYMA is also available in a vial, pre-filled syringe and pre-filled pen for injection under the skin (subcutaneous). Instructions for use for subcutaneous injections in vial, pre-filled syringe and pre-filled pen presentations are provided below.
How to inject STEQEYMA from a vial
- Gather the supplies for the injection.
a. Prepare a clean, flat surface, such as a table or countertop, in a well-lit area.
b. Take the carton containing the STEQEYMA vial that is needed to administer your prescribed dose out of the refrigerator.
c. Make sure you have the following supplies (see Figure A):

- Carton containing the STEQEYMA vial
Not included in the carton:
- Syringe with needle
- 2 alcohol swabs
- Cotton ball or gauze
- Adhesive bandage
- Sharps disposal container
Note: If you don't know the syringe or needle needed contact your healthcare provider for further instructions.
- Check the expiration date on the carton (see Figure B).

- Do not use the medication if the expiration date has passed. If the expiration date has passed, return the entire pack to the pharmacy.
- Wait 30 minutes.
a. Take the carton containing the STEQEYMA vial out of the refrigerator.
b. Leave the carton at room temperature 20°C to 25°C for 30 minutes to allow it to warm up (see Figure C).
- Do not warm the vial using heat sources such as hot water or a microwave.
- If the vial does not reach room temperature, this could cause the injection to feel uncomfortable and make it hard to inject.
- Inspect the STEQEYMA vial.
a. Look at the vial and make sure you have the correct medicine (Steqeyma) and dosage.
b. Look at the vial and make sure it is not cracked or damaged.
c. Do not use STEQEYMA if the vial has been dropped or damaged.
d. Check the expiration date on the label of the vial (see Figure D).
- Do not use STEQEYMA if the expiration date has passed.
- Inspect the medicine.
a. Look at the medicine in the vial and confirm that the liquid is clear to slightly opalescent and colourless to pale yellow. (see Figure E).

- Do not use STEQEYMA if the liquid is discolored or cloudy, or if it contains visible flakes or particles.
- Choose an appropriate injection site (see Figure F).

a. You may inject into:
- the upper thighs.
- the lower abdomen except for the 5 cm around the belly button.
- the outer area of the upper arms if you are a caregiver.
- Do not inject into moles, scares, bruises, or areas where the skin is tender, red, hard or if there are breaks in the skin. If possible, do not use areas of skin that show signs of psoriasis.
- Do not inject through your clothes.
b. Choose a different injection site for each new injection at least 2.5 cm away from the area used for the last injection
- Wash your hands.
a. Wash your hands with soap and water and dry them thoroughly (see Figure G).

- Clean the injection site.
a. Clean the skin with an alcohol swab where you plan to give your injection using a circular motion (see Figure H).
- Let your skin dry before injecting. Do not blow on or touch the injection site again before giving the injection.
- Remove the protective cap from the vial.
a. Remove the protective cap from the STEQEYMA vial (see Figure I).

- Do not remove the rubber stopper.
- Clean the rubber stopper.
a. Wipe the rubber stopper of the vial with an alcohol swab and allow it to dry (see Figure J).

- Do not touch the rubber stopper after you have cleaned it.
b. Place the vial on a flat surface.
- Remove the needle cap from the syringe.
a. Hold the syringe at the middle of the syringe body with the needle pointing away from yourself, and pull the needle cap straight off the syringe (see Figure K).

b. Dispose of the needle cap right away in a sharps disposal container (see Step 19. Dispose of Steqeyma).
- Do not re-cap the syringe.
- Do not touch the needle or let the needle touch anything. Doing so may result in a needle stick injury.
- Do not use the syringe if it has been dropped without the needle cap in place. If this happens call your doctor, nurse or health professional for further instructions.
- Withdraw the correct dose.
a. Put the vial on a flat surface and push the needle through the rubber stopper of the vial.
b. Leave the needle in the vial and turn both the syringe and vial upside down (vial on top).
c. Hold the syringe and vial firmly in one hand. Make sure the tip of the needle is in the liquid.
- It is important that the needle tip is always in the liquid. This stops air bubble from forming in the syringe.
d. With your other hand, pull on the plunger rod to fill the syringe with the amount of liquid prescribed by your doctor (see Figure L).

- Fill the syringe until the black tip of the plunger lines up with the mark that matches your prescribed dose.
For adults and paediatric patients (children and adolescents) 6 years of age and older, who weigh 60 kg or more, pull on the syringe plunger to fill the syringe with the amount of liquid prescribed by your healthcare provider (0.5 mL or 1.0 mL).
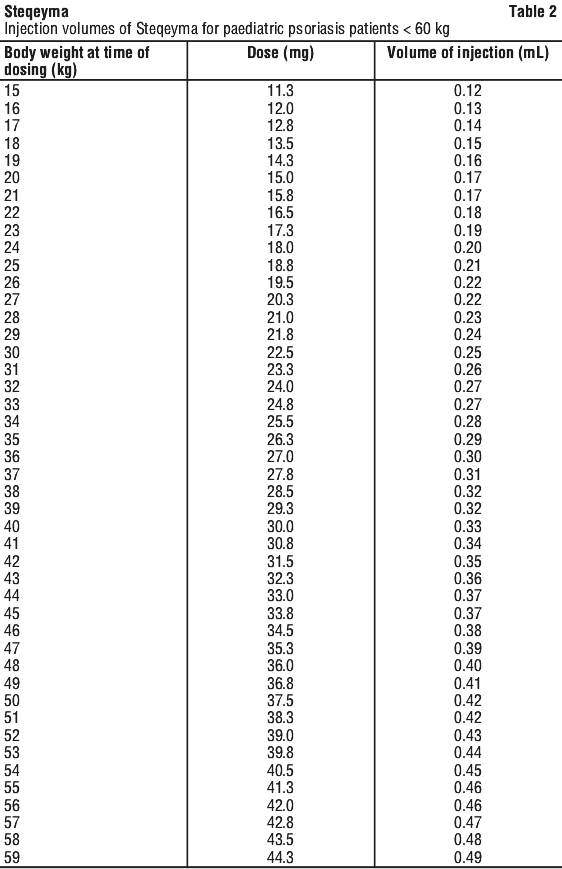
For children and adolescents 6 years of age or older who weigh less than 60 kg, the amount of liquid prescribed by your healthcare provider may be less than 0.5 mL. Your healthcare provider will recommend how much liquid is needed.
- Check for air bubbles.
- Do not remove the needle from the vial.
a. Hold the syringe with the needle pointing up to see if it has any air bubbles inside.
b. If there are air bubbles, gently tap the side of the syringe until the air bubbles go to the top of the syringe (see Figure M).

c. Press the plunger rod until all of the air bubbles (but none of the liquid) have been removed.
- Do not lay the syringe down, or allow the needle to touch anything.
- Insert the needle into the injection site.
a. Hold the body of the syringe in one hand between the thumb and index fingers (see Figure N).

Use the other hand to gently pinch the cleaned skin between your thumb and index finger. Do not squeeze it tightly.
Note: Pinching the skin is important to make sure that you inject under the skin (into the fatty area) but not any deeper (into muscle).
b. With a quick and ‘dart-like motion’, insert the needle completely into the pinched skin at a 45-degree angle (see Figure N).
- Do not pull back on the plunger rod at any time.
- Give the injection.
c. After the needle is inserted use your thumb to slowly and evenly push the plunger rod all the way to the bottom of the syringe body. Keep the skin gently pinched.
- Make sure you have injected all of the STEQEYMA liquid (see Figure O).

- Remove the syringe from the injection site.
a. After the syringe is empty, release the pinched skin and slowly move the syringe away from the injection site (see Figure P).
- Do not re-cap the used needle. Recapping the used needle can lead to a needle-stick injury.
- Do not reuse the syringe.
- Do not rub the injection site.
- Care for the injection site.
- Gently press an alcohol swab over the injection site for a few seconds after the injection. There may be a small amount of blood or liquid at the injection site. This is normal.
If some bleeding occurs, you can press a cotton ball or gauze over the injection site and hold for 10 seconds.
You may cover the injection site with a small adhesive bandage, if necessary.
- If your dose requires 2 injections, IMMEDIATELY give second injection.
- If your dose is 90 mg, you will have two 45 mg vials. You will need to give yourself a second injection right after the first one.
a. Repeat Steps 5-17 for the second injection using a new vial, needle and syringe.
- Choose a different site for the second injection.
- Dispose of STEQEYMA.
a. Put the used syringe in a sharps container right away after use (see Figure Q).

- Do not throw away (dispose of) the syringe in your household trash. If you do not have a sharps disposal container, you may use a household container that is closable and puncture resistant.
- For the safety and health of you and others, needles and used syringes must never be re-used. Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
- Do not throw away any medicines via wastewater or household waste. Ask your pharmacist how tothrow away medicines you no longer use. These measures will help protect the environment.
How to inject STEQEYMA from a pre-filled syringe (45 mg or 90 mg)
- Gather the supplies for the injection
a. Prepare a clean, flat surface, such as a table or counter top, in a well-lit area.
b. Take the carton(s) containing the pre-filled syringe(s) needed to administer your prescribed dose out of the refrigerator.
c. Make sure you have the following supplies (see Figure A)

Carton containing pre-filled syringe
Not included in the carton:
- Cotton ball or gauze
- Adhesive bandage
- Sharps disposal container
- Alcohol swab
- Check the expiration date on the carton (see Figure B).

- Do not use it if the expiration date has passed. If the expiration date has passed, return the entire pack to the pharmacy.
- Wait 30 minutes.
a. Open the carton.
Gripping from the syringe body, lift the pre-filled syringe from the carton.
b. Let the pre-filled syringe stand outside the box for about 30 minutes at room temperature (20°C to 25°C) to allow it to warm up (see Figure C).

- Do not warm the pre-filled syringe using heat sources such as hot water or a microwave.
- This will let the liquid come to a comfortable temperature for injection (room temperature).
- Do not hold by the plunger head, plunger rod, needle guard wings, or needle cover
- Do not pull back on the plunger rod at any time.
- Inspect the pre-filled syringe.
a. Look at the pre-filled syringe and make sure you have the correct medicine (STEQEYMA) and dosage.
b. Check the pre-filled syringe(s) to make sure the number of pre-filled syringes and strength is correct:
- If your dose is 45 mg you will get one 45 mg pre-filled syringe of STEQEYMA.
- If your dose is 90 mg you will get one 90 mg pre-filled syringe of STEQEYMA.
c. Look at the pre-filled syringe and make sure it is not cracked or damaged.
d. Check the expiration date on the label of the pre-filled syringe (see Figure D).

- Do not use if the expiration date has passed.
- Do not shake the pre-filled syringe.
- Inspect the medicine.
a. Look at the medicine and confirm that the liquid is clear to slightly opalescent and colourless to pale yellow. (see Figure E).

- Do not use the pre-filled syringe if the liquid is discoloured or cloudy.
- You may see air bubbles in the liquid. This is normal.
- Choose an appropriate injection site (see Figure F).

a. You may inject into:
- Your upper thighs.
- Your lower abdomen except for the 5 cm around the belly button (navel).
- The outer area of the upper arm if you are a caregiver.
- Do not inject into moles, scars, bruises, or areas where the skin is tender, red, hard, or if there are breaks in the skin. If possible, do not use areas of skin that show signs of psoriasis.
- Do not inject through your clothes.
b. Choose a different injection site for each new injection at least 2.5 cm away from the area used for the last injection.
- Wash your hands.
a. Wash your hands with soap and water and dry them thoroughly (see Figure G).

- Clean the injection site.
a. Clean the injection site with an alcohol swab using a circular motion (see Figure H).

b. Let the skin dry before injecting.
- Do not blow on or touch the injection site again before giving the injection.
- Remove the cap.
a. Remove the needle cover when you are ready to inject your STEQEYMA by holding the body of the pre-filled syringe in one hand between the thumb and index fingers (see Figure I).

- Do not hold the plunger while removing the cap.
- You may notice an air bubble in the pre-filled syringe or a drop of liquid at the tip of needle. This is normal.
b. Dispose of the cap right away in a sharps disposal container (see Step 14 and Figure I).
- Do not use the pre-filled syringe if it is dropped without the needle cover in place. If this happens, please contact your doctor or pharmacist.
- Inject the dose promptly after removing the needle cover.
- Do not re-cap the pre-filled syringe.
- Do not touch the needle. Doing so may result in a needle stick injury.
- Insert the pre-filled syringe into the injection site.
a. Hold the body of the pre-filled syringe in one hand between the thumb and index fingers.
b. Use the other hand to gently pinch the cleaned skin between your thumb and index finger. Do not squeeze it tightly.
Note: Pinching the skin is important to make sure that you inject under the skin (into the fatty area) but not any deeper (into muscle).
c. With a quick and dart-like motion, insert the needle completely into the fold of skin at a 45-degree angle (see Figure J).

- Do not pull back on the plunger rod at any time.
- Give the injection.
a. After the needle is inserted, release the pinch.
b. Slowly push the plunger rod all the way down until the full dose of medicine gets injected, and the syringe is empty (see Figure K).

- Do not change the position of the pre-filled syringe after the injection has started.
- If the plunger rod is not fully pressed, the needle guard will not extend to cover the needle when it is removed.
- Remove the pre-filled syringe from the injection site.
a. After the pre-filled syringe is empty, as the needle is being taken out, slowly remove the needle by lifting your thumb from the plunger rod until the needle is completely covered by the needle guard (see Figure L).

- If the needle is not covered, proceed carefully to dispose of the syringe (see Step 14. Dispose of STEQEYMA).
- Do not reuse the pre-filled syringe.
- Do not rub the injection site.
- Care for the injection site.
a. If some bleeding occurs, treat the injection site by gently pressing, not rubbing, a cotton ball or gauze to the site and apply an adhesive bandage if needed.
- Dispose of STEQEYMA.
a. Put the used pre-filled syringe in a sharps disposal container right away after use (see Figure M).

b. Do not throw away (dispose of) the pre-filled syringe in your household trash. Dispose of sharps in an appropriate container according to your local regulations.
- For the safety and health of you and others, needles and used syringes must never be reused. Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
- Do not throw away any medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help protect the environment.
How to inject STEQEYMA from a pre-filled pen (45 mg or 90 mg)
- Gather the supplies for the injection
a. Prepare a clean, flat surface, such as a table or countertop, in a well-lit area.
b. Remove the 1 carton containing the autoinjector from the refrigerator.
c. Make sure you have the following supplies (see Figure A)

- 1 Autoinjector
Not included in the carton:
- Cotton ball or gauze
- Adhesive bandage
- Sharps disposal container
- Alcohol swab
- Check the expiration date on the carton (see Figure B).

- Do not use it if the expiration date has passed. If the expiration date has passed, return the entire pack to the pharmacy.
- Wait 30 minutes.
a. Open the carton. And remove the autoinjector.
b. Leave the autoinjector at room temperature 68°F to 77°F (20°C to 25°C) for about 30 minutes to allow it to warm up (see Figure C).

- This will let the liquid come to a comfortable temperature for injection (room temperature).
- Do not warm the autoinjector using heat sources such as hot water or a microwave.
- Wash your hands.
a. Wash your hands with soap and water and dry them thoroughly (see Figure D).

- Inspect the autoinjector.
a. Look at the autoinjector and make sure you have the correct medicine (STEQEYMA) and dosage.
b. Look at the autoinjector and make sure it is not cracked or damaged.
c. Check the expiration date on the label of the autoinjector (see Figure E).
- Do not shake the autoinjector.
- Do not use the autoinjector if:
- it has been dropped onto a hard surface.
- it has been cracked or damaged.
- the expiration date has passed.
- Inspect the medicine.
a. Look at the medicine and confirm that the liquid is clear to slightly opalescent and colourless to pale yellow. (see Figure F).

- Do not use the pre-filled syringe if the liquid is discoloured, cloudy, or has large particles
- You may see air bubbles in the liquid. This is normal.
- Choose an appropriate injection site (see Figure G).

a. You may inject into:
- The front of your upper thighs.
- The buttocks
- your stomach (abdomen) except for the 2 inches (5 cm) around the belly button (navel).
- The outer area of the upper arm if you are a caregiver.
- Do not inject into moles, scars, bruises, or areas where the skin is tender, red, hard, or if there are breaks in the skin. If possible, do not use areas of skin that show signs of psoriasis.
- Do not inject through your clothes.
b. Choose a different injection site for each new injection at least 2.5 cm away from the area used for the last injection.
- Clean the injection site.
a. Clean the injection site with an alcohol swab using a circular motion (see Figure H).

b. Let the skin dry before injecting.
- Do not blow on or touch the injection site again before giving the injection.
- Remove the cap.
a. Hold the autoinjector in one hand by the injector body with the cap on top. Gently pull the cap straight off with the other hand (see Figure I).

- Do not remove the cap until you are ready to inject.
- It is normal to see a few drops of liquid come out of the needle.
- Do not touch the needle or needle cover. Doing so may result in a needle stick injury because the needle is inside the needle cover.
- Inject STEQEYMA within 5 minutes of removing the cap.
- Do not re-cap the autoinjector.
b. Dispose of the cap right away in a sharps disposal container (see Step 13 and Figure J).
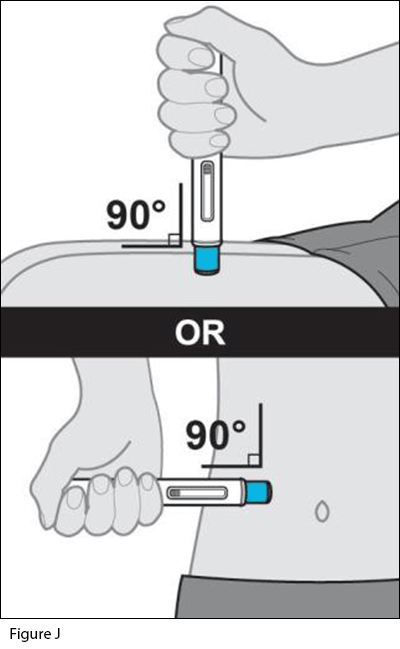
- Place the autoinjector on the injection site.
a. Hold the autoinjector so that you can see the window.
b. Without pinching or stretching the skin, place the autoinjector over the injection site at a 90-degree angle (see Figure J).
- Give the injection.
a. Press the autoinjector firmly against the skin. When the injection starts you will hear the 1st “click” and the light lilac plunger rod will begin to fill the window (see Figure K).
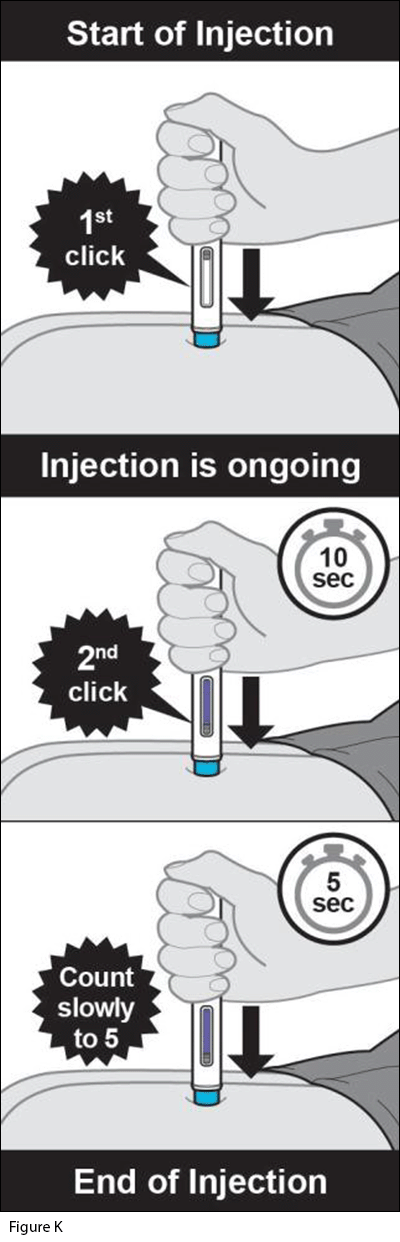
b. Keep holding the autoinjector firmly against the skin and listen for the 2nd “click” (see Figure K).
- Do not change the position of the autoinjector after the injection has started.
c. After you hear the 2nd “click,” continue to hold the autoinjector firmly against the skin and count slowly to 5 to make sure you inject the full dose.
- Remove the autoinjector from the injection site.
a. Look at the autoinjector and make sure that the light lilac plunger rod is filling the window completely.
- You may see the grey stopper in the window. This is normal.
- If the window has not turned completely light lilac or if the medicine is still injecting, this means you have not received a full dose. Call your healthcare provider immediately.
b. Remove the autoinjector from your skin (see Figure L).

- After you remove the autoinjector from the injection site, the needle will be automatically covered (see Figure M).

- Do not reuse the autoinjector.
- Do not rub the injection site.
- Dispose of the autoinjector
a. Throw away the used autoinjector in a sharps disposal container immediately after use (see Figure N).
b. The alcohol swab and packaging may be put in your household trash.
- If you do not have an sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labelled to warn of hazardous waste inside the container.
When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of it.
There may be state or local laws about how you should throw away used needles and syringes. - Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
- Care for the injection site Dispose of STEQEYMA.
a. If some bleeding occurs, treat the injection site by gently pressing, not rubbing, a cotton ball or gauze to the site and apply an adhesive bandage if needed.
If you forget to use STEQEYMA
Make the next injection as soon as you remember, and then continue to use it as you would normally.
Do not take a double dose to make up for the dose you missed.
If you have missed more than one dose, or are not sure what to do, check with your doctor or pharmacist.
If you have trouble remembering when to use your medicine, ask your pharmacist for some hints.
If you use too much STEQEYMA
If you think that you have used too much STEQEYMA, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using STEQEYMA?
Things you should do
- Always follow your doctor's instructions carefully.
- Tell your doctor about your medical conditions before each treatment (see Before you use STEQEYMA).
- Tell your doctor if you become pregnant while using STEQEYMA.
- If you used STEQEYMA while pregnant, tell you baby's doctor about your STEQEYMA use before the baby receives any vaccines, including live vaccines, such as the BCG vaccine (used to prevent tuberculosis), rotavirus vaccine or any other live vaccines.
- If you develop headache, vision problems, seizures or change in mental status (for example confusion), tell your doctor immediately.
- If you are about to start taking a new medicine, tell your doctor and pharmacist that you are using STEQEYMA.
Call your doctor straight away if you:
- get symptoms of an infection, such as a fever, skin sores or feeling tired
- become pregnant while using STEQEYMA. Remind any doctor, dentist or pharmacist you visit that you are using STEQEYMA.
Things you should not do
- Do not stop using this medicine suddenly without checking with your doctor
- You should not receive a live vaccine while taking STEQEYMA.
- Do not use STEQEYMA to treat any other complaint unless your doctor says so.
- Do not give this medicine to anyone else, even if their symptoms seem similar to you.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how STEQEYMA affects you.
Drinking alcohol
Tell your doctor if you drink alcohol.
There is no information on the effects of taking STEQEYMA with alcohol
Looking after your medicine
Follow the instructions in the carton on how to take care of your medicine properly.
Store all presentations of STEQEYMA between 2°C and 8°C in the refrigerator. Do not freeze.
Keep the products in the original cartons to protect from light until the time of use. Do not shake.
STEQEYMA pre-filled syringes and pre-filled pen - room temperature storage:
If needed, you may store STEQEYMA pre-filled syringes and pre-filled pens at room temperature up to 25°C for one period of up to 31 days within the shelf-life. Once the pre-filled syringes and pre-filled pens have been stored at room temperature, they should not be returned to the refrigerator. Discard the pre-filled syringe or pre-filled pen if not used within 31 days at room temperature storage.
- Write the date on the carton, when removed from the refrigerator.
STEQEYMA 45 mg/0.5 mL vial (for subcutaneous use) – room temperature storage:
If needed, you may store STEQEYMA 45 mg/0.5 mL vial at room temperature up to 30°C for one period of up to 15 days.
- Write the date on the carton, when removed from the refrigerator
- Once 45 mg/0.5 mL vial has been stored at room temperature, do not put it back in the refrigerator
- Throw away 45 mg/0.5 mL vial if it has been kept for 15 days and has not been used or reached its original expiry
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
For pre-filled pen only, before use, remove the carton from the refrigerator and keep the pre-filled pen inside the carton and allow to reach room temperature by waiting for 30 minutes.
Keep it where young children cannot reach it.
Heat and dampness can destroy some medicines.
When to discard your medicine
- After injection, used syringes and pens should be placed in a puncture-resistant container, like a sharps container.
- Empty vials, antiseptic wipes, and other supplies can be placed in regular rubbish.
- Dispose of your sharps container according to your state or local regulations.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
Injection site or Infusion-related reactions:
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
Signs of an allergic reaction, such as:
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people. Tell your doctor if you notice any other side effects, particularly if they bother you or do not go away. In general, the side effects of STEQEYMA in children and adolescents 6 years and older are similar to those in adults. Ask your doctor or pharmacist for more information.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What STEQEYMA contains
| Active ingredient (main ingredient) | 45 mg vial or 45 mg, 90 mg pre-filled syringe or pre-filled pen 45 mg ustekinumab 90 mg ustekinumab 130 mg vial 130 mg Ustekinumab |
| Other ingredients (inactive ingredients) | 45 mg vial or 45 mg, 90 mg pre-filled syringe or pre-filled pen - Histidine - Histidine monohydrochloride monohydrate - Sucrose - Polysorbate 80 - Water for injections 130 mg vial - Histidine - Histidine monohydrochloride monohydrate - Methionine - Disodium edetate - Sucrose - Polysorbate 80 - Water for injections |
Do not take this medicine if you are allergic to any of these ingredients.
What STEQEYMA looks like
STEQEYMA injection for subcutaneous use is clear to slightly opalescent, colourless to pale yellow solution and may contain a few small translucent or white particles of protein. This appearance is not unusual for solutions containing protein.
STEQEYMA solution for intravenous use is clear to slightly opalescent, colourless to pale yellow product.
STEQEYMA is available in pack sizes of 1 in the following presentations.
For subcutaneous use:
45 mg ustekinumab in 0.5 mL concentrate solution in a glass vial (AUST R 475406).
45 mg ustekinumab in 0.5 mL solution in a pre-filled syringe (AUST R 420572).
90 mg ustekinumab in 1.0 mL solution in a pre-filled syringe (AUST R 420570).
45 mg ustekinumab in 0.5 mL solution in a pre-filled pen (AUST R 500306).
90 mg ustekinumab in 1.0 mL solution in a pre-filled pen (AUST R 500305).
For intravenous infusion only:
130 mg ustekinumab in 26 mL concentrate solution in a glass vial (AUST R 420571).
Who distributes STEQEYMA
Celltrion Healthcare Australia Pty Ltd.
Suite 13-03 31 Market Street,
Sydney NSW 2000, Australia
Phone: 1800 325 228
This leaflet was prepared in December 2025.
Brand Information
| Brand name | Steqeyma |
| Active ingredient | Ustekinumab |
| Schedule | S4 |
MIMS Revision Date: 01 April 2026
1 Name of Medicine
Ustekinumab.
Steqeyma (Ustekinumab) is a biosimilar medicine to Stelara (Ustekinumab). The evidence for comparability supports the use of Steqeyma for the listed indications.
2 Qualitative and Quantitative Composition
For subcutaneous administration. Steqeyma 45 mg solution for injection in vial. Each vial contains 45 mg ustekinumab in 0.5 mL.
Steqeyma 45 mg solution for injection in pre-filled syringe. Each pre-filled syringe contains 45 mg ustekinumab in 0.5 mL.
Steqeyma 45 mg solution for injection in pre-filled pen. Each pre-filled pen contains 45 mg ustekinumab in 0.5 mL.
Steqeyma 90 mg solution for injection in pre-filled syringe. Each pre-filled syringe contains 90 mg ustekinumab in 1 mL.
Steqeyma 90 mg solution for injection in pre-filled pen. Each pre-filled pen contains 90 mg ustekinumab in 1 mL.
For a full list of excipients, see Section 6.1 List of Excipients.
For intravenous (IV) infusion only. Steqeyma 130 mg concentrate for solution for infusion. Each vial contains 130 mg ustekinumab in 26 mL (5 mg/mL).
For a full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
For subcutaneous administration. Solution for subcutaneous injection.
The solution is clear to slightly opalescent, colourless to pale yellow.
For intravenous (IV) infusion. Concentrate for solution for infusion.
The solution is clear to slightly opalescent, colourless to pale yellow.
4 Clinical Particulars
4.1 Therapeutic Indications
Plaque psoriasis. Adults. Steqeyma is indicated for the treatment of adult patients (18 years or older) with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy.
Paediatric population, 6 years and older. Steqeyma is indicated for the treatment of moderate to severe plaque psoriasis in children and adolescent patients from 6 years of age who are inadequately controlled by, or are intolerant to, other systemic therapies or phototherapies.
Psoriatic arthritis (PsA). Steqeyma, alone or in combination with methotrexate, is indicated for the treatment of signs and symptoms of active psoriatic arthritis in adult patients (18 years and older) where response to previous non-biological DMARD therapy has been inadequate.
Crohn's disease. Steqeyma is indicated for the treatment of adult patients with moderately to severely active Crohn's disease who have had an inadequate response, lost response, or were intolerant to either conventional therapy or a TNFα antagonist or have medical contraindications to such therapies.
Ulcerative colitis. Steqeyma is indicated for the treatment of adult patients with moderately to severely active ulcerative colitis.
4.2 Dose and Method of Administration
Dosing. Plaque psoriasis. Adults. For the treatment of plaque psoriasis, Steqeyma is administered by subcutaneous injection. The recommended dose of Steqeyma is 45 mg administered at weeks 0 and 4, then every 12 weeks thereafter. Alternatively, 90 mg administered over weeks 0 and 4, then every 12 weeks thereafter may be used in patients with a body weight greater than 100 kg.
Dose adjustment. For patients who inadequately respond to dosing every 12 weeks, consideration may be given to treating as often as every 8 weeks. Treatment should be discontinued in patients who have shown no response after 28 weeks of treatment.
Re-treatment. After interruption of therapy, re-treatment with a dosing regimen of weeks 0 and 4, then every 12 weeks thereafter has been shown to be safe and effective.
Paediatric population, 6 years and older. For the treatment of plaque psoriasis, Steqeyma should be administered by subcutaneous injection. The recommended dose of Steqeyma based on body weight is shown in Table 1. Steqeyma should be administered at weeks 0 and 4, then every 12 weeks thereafter. The pre-filled pen is not recommended for use in paediatric patients.
Psoriatic arthritis. For the treatment of psoriatic arthritis, Steqeyma is administered by subcutaneous injection. The recommended dose of Steqeyma is 45 mg administered at weeks 0 and 4, then every 12 weeks thereafter. Some patients with a body weight greater than 100 kg received a 90 mg dose in clinical trials and observed a clinical benefit.
Treatment should be discontinued in patients who have shown no response after 28 weeks of treatment.
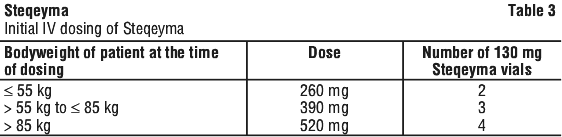
Crohn's disease and ulcerative colitis. For the treatment of Crohn's disease and ulcerative colitis, the recommended treatment regimen is to initiate Steqeyma with a single intravenous (IV) tiered dose based on body weight (Table 3). The infusion solution is to be composed of the number of vials of Steqeyma 130 mg as specified in Table 3.
For some patients, the single IV dose followed by a 90 mg subcutaneous dose 8 weeks later, then every 12 weeks thereafter may be acceptable according to clinical judgment. Patients who inadequately respond to 90 mg subcutaneous dosing every 12 weeks may benefit from an increase in dosing frequency to every 8 weeks (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Immunomodulators and/or corticosteroids may be continued during treatment with Steqeyma. In patients who have responded to treatment with Steqeyma corticosteroids may be reduced or discontinued in accordance with standard of care.
If therapy in Crohn's disease or ulcerative colitis is interrupted, treatment may be resumed with subcutaneous dosing every 8 weeks (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Consideration should be given to discontinuing treatment in patients who show no evidence of therapeutic benefit by week 16.
Use in patients with hepatic or renal impairment. Steqeyma has not been studied in these patient populations. No dose recommendations can be made (see Section 5.2 Pharmacokinetic Properties, Population pharmacokinetic analysis).
Administration. Subcutaneous administration. Steqeyma 45 mg vials, 45 mg and 90 mg pre-filled syringes and pre-filled pens are for subcutaneous injection only. Do not inject into areas where the skin is tender, bruised, red, hard, thick, scaly or affected by psoriasis.
Each vial and pre-filled syringe or pre-filled pen is for single use only and any unused medicinal product should be disposed of in accordance with local requirements.
Steqeyma is intended for use under the guidance and supervision of a health care professional. Patients or their caregivers may inject Steqeyma if a physician determines that it is appropriate and with medical follow-up as necessary, after proper training in subcutaneous injection technique.
In paediatric patients, it is recommended that Steqeyma be administered by a health care provider. The pre-filled pen has not been studied in the paediatric population and it not recommended for use in paediatric patient.
Comprehensive instructions for the subcutaneous administration of Steqeyma are given in the Consumer Medicine Information for the pre-filled syringe and pre-filled pen. Patients should be instructed to inject the full amount of Steqeyma subcutaneously according to the directions provided in the Consumer Medicine Information.
Vials. When using the single-use vial, a 27 gauge, ½ inch needle is recommended.
Inspect Steqeyma visually for particulate matter and discolouration prior to administration. Steqeyma is a colourless to light yellow solution and may contain a few small translucent or white particles. Do not use Steqeyma if it is discoloured or cloudy, or if other particulate matter is present. Steqeyma does not contain preservatives; therefore, discard any unused product remaining in the vial, pre-filled syringe or pre-filled penin accordance with local requirements and must never be re-used.
Following administration of Steqeyma, the syringe should be disposed of in accordance with accepted medical practices for used syringes.
Pre-filled syringe or pre-filled pen. Each pre-filled syringe and pre-filled pen is for single dose only. Patients may encounter resistance while injecting. It is important to instruct patients to inject the full amount to receive either 45 mg or 90 mg of Steqeyma.
For pre-filled pen only, before injection, remove Steqeyma from the refrigerator and allow it to reach room temperature (30 minutes) inside the carton without removing the pre-filled pen or needle cap.
Intravenous infusion (Crohn's disease and ulcerative colitis). Steqeyma 130 mg vial is for IV infusion only.
Instructions for dilution and IV infusion of Steqeyma 130 mg for IV infusion (Crohn's disease and ulcerative colitis). Steqeyma 130 mg solution must be diluted and prepared for IV infusion by a healthcare professional using aseptic technique. The IV infusion should be administered by qualified healthcare professionals.
1. Calculate the dose and the number of Steqeyma vials needed based on patient's body weight (see Table 3). Each 26 mL vial of Steqeyma contains 130 mg of ustekinumab.
2. Withdraw and then discard a volume of the 0.9% w/v sodium chloride solution from the 250 mL infusion bag equal to the volume of Steqeyma to be added. (Discard 26 mL sodium chloride for each vial of Steqeyma needed, for 2 vials-discard 52 mL, for 3 vials-discard 78 mL, for 4 vials-discard 104 mL).
3. Withdraw 26 mL of Steqeyma from each vial needed and add it to the 250 mL infusion bag. The final volume in the infusion bag should be 250 mL. Gently invert or swirl the bag to mix the solution. Do not shake.
4. Once diluted, the infusion solution may be stored for up to eight hours prior to infusion.
5. Visually inspect the diluted solution before administration. Do not use if visibly opaque particles, discoloration or foreign particles are observed.
6. Administer the diluted solution over a period of at least one hour.
7. Use only an infusion set with an in-line, sterile, non-pyrogenic, low protein-binding filter (pore size 0.2 micrometer).
8. Do not infuse Steqeyma concomitantly in the same intravenous line with other agents.
9. Each vial is for single use only and any unused medicinal product should be disposed of in accordance with local requirements.
4.3 Contraindications
Severe hypersensitivity to ustekinumab or to any of the excipients (see Section 6.1 List of Excipients).
Steqeyma should not be given to patients with a clinically important, active infection (see Section 4.4 Special Warnings and Precautions for Use).
4.4 Special Warnings and Precautions for Use
Serious infections. Ustekinumab is a selective immunosuppressant and may have the potential to increase the risk of infections and reactivate latent infections.
In clinical studies, serious bacterial, fungal, and viral infections have been observed in patients receiving ustekinumab. Caution should be exercised when considering the use of ustekinumab in patients with a chronic infection or a history of recurrent infection.
Prior to initiating treatment with ustekinumab, patients should be evaluated for tuberculosis infection. Ustekinumab should not be given to patients with active tuberculosis. Treatment of latent tuberculosis infection should be initiated prior to administering ustekinumab. Anti-tuberculosis therapy should also be considered prior to initiation of ustekinumab in patients with a past history of latent or active tuberculosis in whom an adequate course of treatment cannot be confirmed. Patients receiving ustekinumab should be monitored closely for signs and symptoms of active tuberculosis during and after treatment.
Patients should be instructed to seek medical advice if signs or symptoms suggestive of an infection occur. If a patient develops a serious infection they should be closely monitored and ustekinumab should not be administered until the infection resolves (see Section 4.8 Adverse Effects (Undesirable Effects)).
Non-infectious pneumonia. Cases of interstitial pneumonia, eosinophilic pneumonia and cryptogenic organizing pneumonia have been reported during post-approval use of ustekinumab. Clinical presentations included cough, dyspnoea, and interstitial infiltrates following one to three doses. Serious outcomes have included respiratory failure and prolonged hospitalization. Patients improved with discontinuation of therapy and in certain cases administration of corticosteroids. If diagnosis is confirmed, discontinue ustekinumab and institute appropriate treatment.
Malignancies. Ustekinumab is a selective immunosuppressant. Immunosuppressive agents have the potential to increase the risk of malignancy. Some patients who received ustekinumab in clinical studies developed cutaneous and non-cutaneous malignancies (see Section 4.8 Adverse Effects (Undesirable Effects)).
Ustekinumab has not been studied in patients with a history of malignancy. Caution should be exercised when considering the use of ustekinumab in patients with a history of malignancy or when considering continuing treatment in patients who develop a malignancy.
All patients, in particular those greater than 60 years of age, patients with a medical history of prolonged immunosuppressant therapy or those with a history of PUVA treatment, should be monitored for the appearance of non-melanoma skin cancer (see Section 4.8 Adverse Effects (Undesirable Effects)).
Hypersensitivity and infusion-related reactions. In post-marketing experience, serious hypersensitivity reactions, including anaphylaxis and angioedema, have been reported. Infusion-related reactions were observed in clinical trials (see Section 4.8 Adverse Effects (Undesirable Effects)). Serious infusion-related reactions including anaphylactic reactions to the infusion have been reported in the post-marketing setting.
If an anaphylactic or other serious hypersensitivity or infusion-related reaction occurs, appropriate therapy should be instituted and administration of ustekinumab should be discontinued immediately (see Section 4.8 Adverse Effects (Undesirable Effects)).
Immunisations. It is recommended that live viral or live bacterial vaccines (such as Bacillus of Calmette and Guérin [BCG]) not be given concurrently with ustekinumab.
Before live viral or live bacterial vaccination, treatment with ustekinumab should be withheld for at least 15 weeks after the last dose and can be resumed at least 2 weeks after vaccination. Prescribers should consult the Product Information for the specific vaccine for additional information and guidance on concomitant use of immunosuppressive agents post vaccination and considering the benefit risk of ustekinumab treatment in the patient.
No data are available on the secondary transmission of infection by live vaccines in patients receiving ustekinumab. Caution is advised when administering some live vaccines to household contacts of patients receiving ustekinumab because of the potential risk for shedding from the household contact and transmission to the patient.
Patients receiving ustekinumab may receive concurrent inactivated or non-live vaccinations.
Long term treatment with ustekinumab does not suppress the humoral immune response to pneumococcal polysaccharide or tetanus vaccines (see Section 5.1 Pharmacodynamic Properties).
Infant exposure in utero. For infants exposed in utero to ustekinumab, a six month waiting period following birth is recommended before the administration of live vaccines. Administration of a live vaccine prior to 6 months of age may be considered if ustekinumab serum levels are undetectable in the infant and the benefit of the vaccination clearly outweighs the theoretical risk of administration of live vaccines to the infant (see Section 4.6 Fertility, Pregnancy and Lactation).
Immunosuppression. In psoriasis studies, the safety and efficacy of ustekinumab in combination with immunosuppressive agents or phototherapy have not been evaluated. In psoriatic arthritis studies, concomitant methotrexate (MTX) use did not appear to influence the safety or efficacy of ustekinumab. In Crohn's disease and ulcerative colitis studies, concomitant use of immunomodulators (6-mercaptopurine (6-MP), azathioprine (AZA), MTX) or corticosteroids did not appear to influence the safety or efficacy of ustekinumab. Caution should be exercised when considering concomitant use of immunosuppressive agents and ustekinumab or when transitioning from other biologic agents.
Immunotherapy. Ustekinumab has not been evaluated in patients who have undergone allergy immunotherapy. Ustekinumab may affect allergy immunotherapy. Caution should be exercised in patients receiving or who have received allergy immunotherapy particularly for anaphylaxis.
Posterior reversible encephalopathy syndrome (PRES). Posterior reversible encephalopathy syndrome (PRES), also known as Reversible Posterior Leukoencephalopathy Syndrome (RPLS), is a neurological disorder, which is not caused by demyelination or a known infectious agent. Conditions with which it has been associated include preeclampsia, eclampsia, acute hypertension, cytotoxic agents and immunosuppressive therapy. Fatal outcomes have been reported in this condition.
Two cases of PRES were reported in clinical trials. Cases have also been reported in postmarketing experience in patients with psoriasis, psoriatic arthritis and Crohn's disease. Clinical presentation included headaches, seizures, confusion, visual disturbances, and imaging changes consistent with PRES a few days to several months after ustekinumab initiation. A few cases reported latency of a year or longer. Patients recovered with supportive care following withdrawal of ustekinumab. Monitor all patients treated with ustekinumab for signs and symptoms of PRES.
If PRES is suspected, promptly administer appropriate treatment and discontinue ustekinumab.
Serious skin conditions. In patients with psoriasis, exfoliative dermatitis has been reported following ustekinumab treatment. Patients with plaque psoriasis may develop erythrodermic psoriasis, with symptoms that may be clinically indistinguishable from exfoliative dermatitis, as part of the natural course of their disease. As part of the monitoring of the patient's psoriasis, physicians should be alert for symptoms of erythrodermic psoriasis or exfoliative dermatitis. If these symptoms occur, appropriate therapy should be instituted. Ustekinumab should be discontinued if a drug reaction is suspected.
Use in the elderly. Of the 6709 patients exposed to ustekinumab, a total of 353 were 65 years or older (183 patients with psoriasis, 69 patients with psoriatic arthritis, 58 with Crohn's disease and 43 patients with ulcerative colitis). No major age-related differences in clearance or volume of distribution were observed in clinical studies. Although no overall differences in efficacy or safety were observed between older and younger patients in clinical studies in approved indications, the number of patients aged 65 and over is not sufficient to determine whether they respond differently from younger patients.
Paediatric use. Specific studies of ustekinumab in paediatric patients below 6 years of age have not been conducted. The pharmacokinetics of ustekinumab in paediatric psoriasis patients, 6 to 17 years of age, treated with the recommended dose was generally comparable to that in the adult psoriasis population. No pharmacokinetic, safety or efficacy data are available in paediatric patients with psoriatic arthritis, Crohn's disease or ulcerative colitis.
Effects on laboratory tests. No data available.
4.5 Interactions with Other Medicines and Other Forms of Interactions
Specific drug interaction studies have not been conducted with ustekinumab (see Section 5.2 Pharmacokinetic Properties).
Live vaccines should not be given concurrently with ustekinumab. Recommendations for infants exposed to ustekinumab in utero are provided (see Section 4.4 Special Warnings and Precautions for Use, Immunisations).
CYP450 substrates. The formation of CYP450 enzymes can be altered by increased levels of certain cytokines (e.g. IL-1, IL-6, IL-10, TNFα, IFN) during chronic inflammation. Thus, ustekinumab, an antagonist of IL-12 and IL-23, could normalize the formation of CYP450 enzymes. Upon initiation of ustekinumab in patients who are receiving concomitant CYP450 substrates, particularly those with a narrow therapeutic index, monitoring for therapeutic effect (e.g. for warfarin) or drug concentration (e.g. for ciclosporin) should be considered and the individual dose of the drug adjusted as needed.
The effects of IL-12 or IL-23 on the regulation of CYP450 enzymes were evaluated in an in vitro study using human hepatocytes, which showed that IL-12 and/or IL-23 at levels of 10 nanogram/mL did not alter human CYP450 enzyme activities (CYP1A2, 2B6, 2C9, 2C19, 2D6, or 3A4). However, the clinical relevance of this in vitro data has not been established.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. In a male fertility study in cynomolgus monkeys, no ustekinumab-related effects on mating behaviour, sperm parameters, or serum concentrations of male hormones were observed following twice weekly subcutaneous administration of ustekinumab at doses up to 45 mg/kg.
The effect of ustekinumab on female fertility has not been evaluated. A female fertility toxicity study was conducted in mice using an analogous antibody that binds to and inhibits IL-12 and IL-23 activity in mice. Twice weekly subcutaneous administration of the anti-mouse IL-12/23 antibody was well tolerated at doses up to 50 mg/kg and no adverse effects on female fertility parameters were observed.
Use in pregnancy. (Category B1)
It is not known whether ustekinumab can cause foetal harm when administered to a pregnant woman or can affect reproduction capacity. Ustekinumab should be given to a pregnant woman only if the benefit clearly outweighs the risk.
Women of childbearing potential should use effective methods of contraception during treatment and for at least 15 weeks after treatment.
Developmental toxicity studies of ustekinumab were conducted in cynomolgus monkeys. No evidence of maternal toxicity, embryotoxicity or teratogenicity was observed at doses up to 45 mg/kg following weekly or twice weekly administration via the IV or SC routes, respectively, during the period of organogenesis. However, animal reproductive and developmental studies are not always predictive of human response.
Use in lactation. Limited data from published literature suggests that ustekinumab is excreted in human breast milk in very small amounts. While systemic exposure to a breastfed infant is expected to be low because ustekinumab is a large molecule and is likely degraded in the gastrointestinal tract, it is not known if ustekinumab is absorbed systemically after ingestion. Because of the potential for adverse reactions in nursing infants from ustekinumab, a decision on whether to discontinue breast-feeding during treatment and up to 15 weeks after treatment or to discontinue therapy with ustekinumab must be made, taking into account the benefit of breast-feeding to the child and the benefit of ustekinumab therapy to the woman.
Maternal treatment of monkeys with ustekinumab at doses up to 45 mg/kg twice weekly SC from gestation Day 20 to post-partum Day 33 had no adverse effects on offspring development. However, animal reproductive and developmental studies are not always predictive of human response.
4.7 Effects on Ability to Drive and Use Machines
No studies on the effects on the ability to drive and use machines have been performed.
4.8 Adverse Effects (Undesirable Effects)
Clinical studies experience in adult patients with psoriasis, psoriatic arthritis, Crohn's disease and ulcerative colitis. The safety data described in Table 4 reflect exposure to ustekinumab in 14 Phase 2 and Phase 3 studies in 6709 patients (4135 with psoriasis and/or psoriatic arthritis, 1749 for Crohn's disease, and 825 with ulcerative colitis in UC-1 and UC-2 clinical trials), with duration of exposure to ustekinumab presented in Table 4.

Table 5 provides a summary of adverse drug reactions from the clinical studies. The frequency of these adverse reactions was based on those that occurred during the initial controlled periods of the clinical studies. The adverse drug reactions are ranked by frequency, using the following convention:
Very common (> 1/10), common (frequent) (> 1/100, < 1/10), uncommon (infrequent) (> 1/1,000, < 1/100), rare (> 1/10,000, < 1/1,000).
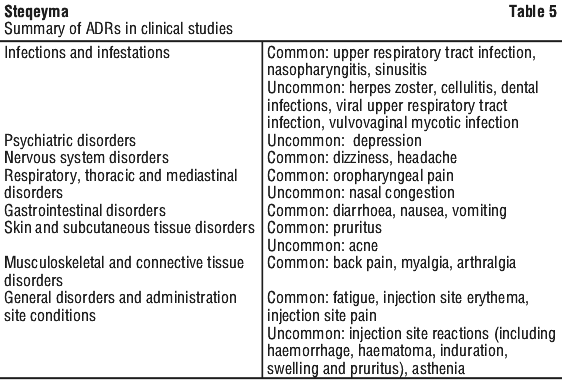
In the controlled and non-controlled portions of psoriasis, psoriatic arthritis, Crohn's disease and ulcerative clinical studies representing 11581 patient-years of exposure in 6709 patients, the median follow-up was 1.0 years; 1.1 years for psoriatic disease studies, 0.6 year for Crohn's disease studies and 1.0 years for ulcerative colitis studies. The rate of infection was 0.91 per patient-year of follow-up in ustekinumab-treated patients. The incidence of serious infections was 0.02 per patient-year of follow-up in ustekinumab-treated patients (199 serious infections in 11581 patient-years of follow-up) and included pneumonia, anal abscess, cellulitis, diverticulitis, osteomyelitis, viral infections, gastroenteritis, and urinary tract infections.
In clinical studies, patients with latent tuberculosis who were concurrently treated with isoniazid did not develop tuberculosis. One case of tuberculosis reactivation occurred in a subject with abnormal baseline chest X-Ray and without treatment for latent TB while on ustekinumab therapy. The subject fully recovered with appropriate treatment.
Malignancy. In the placebo-controlled period of the psoriasis, psoriatic arthritis, Crohn's disease and ulcerative colitis clinical studies, the incidence of malignancies excluding non-melanoma skin cancer was 0.11 per 100 patient-years of follow-up for ustekinumab-treated patients (1 patient in 929 patient-years of follow-up) compared with 0.23 per 100 patient-years of follow-up for placebo-treated patients (1 patient in 434 patient-years of follow-up).
The incidence of non-melanoma skin cancer was 0.43 per 100 patient-years of follow-up for ustekinumab-treated patients (4 patients in 929 patient-years of follow-up) compared with 0.46 per 100 patient-years of follow-up for placebo-treated patients (2 patients in 433 patient-years of follow-up).
In the controlled and non-controlled periods of the psoriasis, psoriatic arthritis, Crohn's disease and ulcerative colitis clinical studies representing 11561 patient-years of exposure in 6709 patients, the median follow-up was 1.0 years; 1.1 years for psoriatic disease studies, 0.6 years for Crohn's disease studies, and 1.0 years for ulcerative colitis studies. Malignancies excluding non-melanoma skin cancers were reported in 62 patients in 11561 patient-years of follow-up (incidence of 0.54 per 100 patient-years of follow-up for ustekinumab-treated patients). The rate of malignancies reported in ustekinumab-treated patients was comparable to the rate expected in the general population (standardized incidence ratio = 0.93 [95% confidence interval:0.71,1.20]). The most frequently observed malignancies, other than non-melanoma skin cancer, were prostate, colorectal, melanoma in situ and breast. The incidence of non-melanoma skin cancer was 0.49 per 100 patient-years of follow-up for ustekinumab-treated patients (56 patients in 11545 patient-years of follow-up). The ratio of patients with basal versus squamous cell skin cancers (3:1) is comparable with the ratio expected in the general population (see Section 4.4 Special Warnings and Precautions for Use).
Hypersensitivity and infusion-related reactions. Subcutaneous administration. During the controlled periods of the psoriasis and psoriatic arthritis clinical studies of ustekinumab, rash and urticaria have each been observed in < 1% of patients.
IV administration. In Crohn's disease and ulcerative colitis IV induction studies, no events of anaphylaxis or other serious infusion reactions with ustekinumab were reported. In these studies, 2.2% of 785 placebo treated patients and 1.9% of 790 patients treated with the recommended dose of ustekinumab reported adverse events occurring during or within an hour of the infusion. Serious infusion-related reactions including anaphylactic reactions have been reported in the post-marketing setting (see Section 4.4 Special Warnings and Precautions for Use).
Immunogenicity. In psoriasis and psoriatic arthritis clinical studies, up to 12.4% of patients treated with ustekinumab developed antibodies to ustekinumab. Patients positive for antibodies to ustekinumab tended to have lower efficacy, however, antibody positivity did not preclude a clinical response. In psoriasis studies, the majority of patients who were positive for antibodies to ustekinumab had neutralising antibodies.
In Crohn's disease and ulcerative colitis clinical studies, 2.9 and 4.6% of patients, respectively, developed antibodies to ustekinumab when treated with ustekinumab for approximately one year. No apparent association between the development of antibodies to ustekinumab and the development of injection site reactions was observed.
Comparability immunogenicity of Steqeyma and Stelara. The immunogenicity of CT-P43 was evaluated in healthy male subjects in Studies CT-P43 1.1 and CT-P43 1.2 and plaque psoriasis patients in Study CT-P43 3.1 using the state-of-the-art and validated immunogenicity assays in accordance with the current international regulatory guidelines.
The proportions of subjects who had at least 1 post-treatment ADA positive result and NAb positive result showed that CT-P43 appeared to be less immunogenic than the reference products in both study with healthy subjects (Study 1.1 and 1.2) and the patients with plaque psoriasis (Study 3.1). Although CT-P43 showed lower immunogenicity results compared to Stelara, the impact of ADA incidence and titer of CT-P43 on the clinical outcome was similar to that of Stelara. Development of antibodies to ustekinumab were associated with reduced serum concentrations but have no significant impact on efficacy and safety.
Clinical studies experience in paediatric patients with psoriasis. The safety of ustekinumab has been studied in two phase 3 studies of paediatric patients with moderate to severe plaque psoriasis. The first study was in 110 patients from 12 to 17 years of age treated for up to 60 weeks (CADMUS) and the second study was in 44 patients from 6 to 11 years of age treated for up to 56 weeks (CADMUS Jr.). In general, the adverse events reported in these two studies with safety data up to 1 year were similar to those seen in previous studies in adults with plaque psoriasis. (see Clinical studies experience in adult patients with psoriasis, psoriatic arthritis, Crohn's disease and ulcerative colitis section above).
Adverse events. The following adverse events have been reported in patients treated with ustekinumab. A causal relationship to ustekinumab is uncertain.
In psoriasis clinical trials of ustekinumab, serious cardiovascular events, including cardiovascular death, myocardial infarction, and stroke, were reported in 0.3% of patients who received ustekinumab compared with 0% of patients treated with placebo, during the placebo-controlled period. Individuals with chronic inflammatory diseases, such as psoriasis, have higher rates of cardiovascular risk factors and cardiovascular events. Rates of myocardial infarction and stroke reported in ustekinumab - treated patients were comparable to rates expected in the general population.
In clinical trials for Crohn's disease, there is no consistent evidence that ustekinumab increases cardiovascular risk in patients treated with ustekinumab through approximately 1 year of treatment. Results from the Crohn's disease studies, up to 1 year, did not change the previous assessment of the impact of ustekinumab on serious major adverse cardiovascular event (MACE).
Adverse events of depression were reported in some patients who received ustekinumab in psoriasis clinical trials, including rare events of suicidality. Individuals with psoriasis have higher rates of depression, and it is not known if ustekinumab may have contributed to these events since ustekinumab also resulted in improvements of the Hospital Anxiety and Depression Scale (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Comparability of Steqeyma and Stelara adverse events. There were no notable differences in the incidence or nature of Adverse Events (AEs) between the CT-P43 and Stelara treatment groups across Studies CT-P43 3.1, CT-P43 1.2 and CT-P43 1.1, and the safety profile of each treatment group was in line with the known safety profile of Stelara. As a result, the analysis of AEs obtained in the clinical studies with CT-P43 illustrated that the benefit-risk profile of CT-P43 is similar to that of Stelara.
Post-marketing data. The adverse drug reactions in Table 6 are ranked by frequency* using the following convention: Very common ≥ 1/10; common ≥ 1/100 and < 1/10; uncommon ≥ 1/1,000 and < 1/100; rare ≥ 1/10,000 and < 1/1,000; very rare < 1/10,000, including isolated reports.

There have been reports of rapidly growing and/or multiple squamous cell carcinomas of the skin in patients receiving ustekinumab who had multiple pre-existing risk factors for developing non-melanoma skin cancer. A causal relationship of these adverse events to ustekinumab is uncertain.
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
4.9 Overdose
Single doses up to 6 mg/kg intravenously have been administered in clinical studies without dose-limiting toxicity. In case of overdose, it is recommended that the patient be monitored for any signs or symptoms of adverse reactions or effects and appropriate symptomatic treatment be instituted immediately.
For information on the management of overdose please contact the Poison Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action. Ustekinumab is a human IgG1 kappa monoclonal antibody that specifically binds to the shared p40 protein subunit of the human cytokines interleukin (IL)-12 and IL-23. Ustekinumab inhibits the bioactivity of human IL-12 and IL-23 by preventing p40 from binding to the IL-12Rbeta1 receptor protein expressed on the surface of immune cells. Ustekinumab cannot bind to IL-12 or IL-23 that is already bound to IL-12Rbeta1 cell surface receptors. Thus, ustekinumab is not expected to contribute to complement- or antibody-mediated cytotoxicity of cells with IL-12 and/or IL-23 receptors.
IL-12 and IL-23 are heterodimeric cytokines secreted by activated antigen presenting cells, such as macrophages and dendritic cells. IL-12 stimulates natural killer (NK) cells and drives the differentiation of CD4+ T cells toward the T helper 1 (Th1) phenotype and stimulates interferon gamma (IFNγ) production. IL-23 induces the T helper 17 (Th17) pathway and promotes secretion of IL-17A, IL-21, and IL-22. Levels of IL-12 and IL-23 are elevated in the skin and blood of patients with psoriasis, and serum IL12/23p40 distinguishes patients with psoriatic arthritis from healthy individuals, implicating IL-12 and IL-23 in the pathophysiology of psoriatic inflammatory diseases. Genetic polymorphisms in IL23A, IL23R and IL-12B genes confer susceptibility to these disorders. IL-12 and IL-23 are highly expressed in lesional psoriatic skin, and IL-12-mediated induction of IFNγ correlates with psoriasis disease activity. IL-23 responsive T-cells have been found in the enthuses in a mouse model of inflammatory arthritis, where IL-23 drives entheseal inflammation. In addition, there is pre-clinical evidence implicating IL-23 and downstream pathways in bone erosion and destruction through up-regulation of receptor activator of nuclear factor κB ligand (RANKL), which activates osteoclasts.
In patients with Crohn's disease, IL-12 and IL-23 are elevated in the intestines and lymph nodes. This is accompanied by increases in serum IFNγ and IL-17A levels, suggesting that IL-12 and IL-23 promote Th1 and Th17 activation in Crohn's disease. Both IL-12 and IL-23 can also stimulate TNFα production by T cells, resulting in chronic intestinal inflammation and epithelial cell injury. Significant associations have been found between Crohn's disease and genetic polymorphisms in the IL23R and IL12B genes, suggesting a potential causal role for IL-12/23 signaling in the disease. This is supported by pre-clinical data demonstrating that IL-12/23 signaling is required for intestinal injury in mouse models of inflammatory bowel disease.
By binding the shared p40 subunit of IL-12 and IL-23, ustekinumab may exert its clinical effects in psoriasis, psoriatic arthritis, Crohn's disease and ulcerative colitis through interruption of the Th1 and Th17 cytokine pathways, which are central to the pathology of these diseases.
Pharmacodynamics. Treatment with ustekinumab resulted in significant improvement in histological measures of psoriasis including epidermal hyperplasia and cell proliferation. These results are consistent with the clinical efficacy observed.
In patients with psoriasis and/or psoriatic arthritis, ustekinumab had no apparent effect on the percentages of circulating immune cell populations including memory and naive T cell subsets or circulating cytokine levels. Systemic markers of inflammation were measurable in the serum at baseline and 4 markers (MDC, VEGF, MCSF-1 and YKL-40) showed modest differences in concentration post-treatment in ustekinumab-treated patients as compared to placebo.
In psoriasis and psoriatic arthritis studies, clinical response (improvement in Psoriasis Area and Severity Index [PASI] or ACR measurements, respectively) appeared to be related to serum ustekinumab levels. Patients with psoriasis with PASI response had higher median serum concentrations of ustekinumab than those with lower clinical responses. In psoriasis studies, the proportion of patients with psoriasis who achieved PASI 75 response increased with increasing serum levels of ustekinumab. The proportion of patients who achieved PASI 75 response at Week 28 increased with increasing serum ustekinumab trough levels at Week 28. In psoriatic arthritis studies, patients achieving an ACR 20 response had higher median serum concentrations of ustekinumab than ACR 20 non-responders. The proportion of patients who achieved ACR 20 and ACR 50 response increased with increasing serum levels of ustekinumab.
In patients with Crohn's disease, treatment with ustekinumab resulted in a significant decrease in inflammatory markers including C-Reactive Protein (CRP) and faecal calprotectin. CRP was assessed during the study extension and the reductions observed during maintenance were generally sustained through week 252. Reductions in serum IFNγ and IL-17A, which are IL-12 and IL-23 regulated pro-inflammatory cytokines, were achieved and maintained in ustekinumab treated patients through Week 44 compared to placebo (52 weeks since the first dose of ustekinumab). At week 6, expression of genes such as IL-12Rβ1 and IL-23 were reduced in inflamed colon tissue from Crohn's disease patients, who were responders to ustekinumab treatment while no significant changes were observed in placebo treated patients.
In patients with ulcerative colitis, treatment with ustekinumab resulted in a decrease in inflammatory markers including CRP and faecal calprotectin during the induction phase, which were maintained throughout the maintenance phase and study extension through week 200.
Immunisation. During the long-term extension of a phase 3 psoriasis study (PHOENIX 2), patients treated with ustekinumab for at least 3.5 years mounted similar antibody responses to both pneumococcal polysaccharide and tetanus vaccines as a non-systemically treated psoriasis control group. Similar proportions of patients developed protective levels of anti-pneumococcal and anti-tetanus antibodies and antibody titres were similar among ustekinumab-treated and control patients.
Comparability of pharmacodynamic properties of Steqeyma and Stelara. In vitro pharmacodynamic studies to compare interleukin (IL)-12/23 p40 binding affinity (enzyme-linked immunosorbent assay [ELISA]), cell-based human IL-12/23 binding affinity (cell-based enzyme-linked immunosorbent assay [CELISA]), complement component 1q (C1q) binding affinity (ELISA), Fc receptor (FcγRIIIa [V-type], FcγRIIIb, FcγRIIa, FcγRIIb, FcγRI and FcRn) binding affinity (surface plasmon resonance [SPR]), antibody-dependent cellular cytotoxicity (ADCC) activity and complement-dependent cytotoxicity (CDC) activity have been performed to demonstrate similarity in the mechanism of action between Steqeyma and Stelara. The outcome of the similarity tests showed that all Steqeyma batches demonstrated comparable properties with respect to the biological activity when compared to Stelara.
Clinical trials. Plaque psoriasis (adults). The safety and efficacy of ustekinumab was assessed in 2 Phase 3 studies (A Phase 3 multicentre, randomised, double-blind, placebo-controlled trial evaluating the efficacy and safety of CNTO 1275 in the treatment of subjects with moderate to severe plaque-type psoriasis followed by long-term extension [PHOENIX] 1 and PHOENIX 2). A total of 1996 patients were enrolled in these studies.
The safety and efficacy of ustekinumab have not been established beyond 4 years.
The studies enrolled adults (≥ 18 years) with chronic (> 6 months) plaque psoriasis who had a minimum body surface area (BSA) involvement of 10%, and PASI score ≥ 12 and who were candidates for systemic therapy or phototherapy. Patients with guttate, erythrodermic, or pustular psoriasis were excluded from the studies. No concomitant anti-psoriatic therapies were allowed during the study with the exception of low-potency topical corticosteroids on the face and groin after week 12.
The Psoriasis Area and Severity Index (PASI) is a composite score that assesses the fraction of body surface area involved with psoriasis and the severity of psoriatic changes within the affected regions (plaque thickness/induration, erythema, and scaling). PASI numeric scores range from 0 to 72, with higher scores representing more severe disease.
Patients achieving ≥ 75% improvement in PASI from baseline (PASI 75) were considered PASI 75 responders. Patients originally randomised to ustekinumab who were PASI 75 responders at both weeks 28 and 40 were considered long-term PASI 75 responders. Patients achieving ≥ 90% improvement in PASI from baseline (PASI 90) were considered PASI 90 responders and patients with ≥ 50% improvement in PASI from baseline (PASI 50) were considered PASI 50 responders. Patients who achieved ≥ 50% but less than 75% improvement in PASI from baseline were considered partial responders. Patients with < 50% improvement in PASI from baseline were considered non-responders.
Other key efficacy assessments included:
The Physician's Global Assessment (PGA), a 6-category scale focusing on plaque thickness/induration, erythema, and scaling.
The Dermatology Life Quality Index (DLQI), a dermatology-specific quality of life instrument, with a lower score indicating an improved quality of life.
The SF-36, a health survey questionnaire consisting of multi-item scales measuring 8 health concepts (PHOENIX 1 only).
The Nail Psoriasis Severity Index (NAPSI), a physician-assessed score that measures the severity of nail involvement (PHOENIX 1 only).
The Hospital Anxiety and Depression Scale (HADS), a self-rating tool developed to evaluate psychological measures in patients with physical ailments (PHOENIX 2 only).
The Work Limitations Questionnaire (WLQ), a 25-item, self-administered questionnaire that was used to measure the impact of chronic health conditions on job performance and work productivity among employed populations (PHOENIX 2 only).
The Itch Visual Analogue Scale, (Itch VAS) used to assess the severity of itch at the time of the assessment (PHOENIX 1 only).
PHOENIX 1. PHOENIX 1 evaluated the safety and efficacy of ustekinumab versus placebo in 766 patients with plaque psoriasis and the efficacy of every 12 week dosing for patients who were PASI 75 responders. Patients randomised to ustekinumab received 45 mg or 90 mg doses at weeks 0 and 4 followed by the same doses every 12 weeks. Patients randomised to receive placebo at weeks 0 and 4 crossed over to receive ustekinumab (either 45 mg or 90 mg) at Weeks 12 and 16 followed by the same dose every 12 weeks.
Maintenance dosing (every 12 weeks). To evaluate the therapeutic benefit of maintenance dosing with ustekinumab, patients originally randomised to ustekinumab who were PASI 75 responders at both weeks 28 and 40 were re-randomised to either maintenance dosing of ustekinumab every 12 weeks or to placebo (i.e. withdrawal of therapy). Patients who were re-randomised to placebo at week 40 reinitiated ustekinumab at their original dosing regimen when they experienced at least a 50% loss of their PASI improvement obtained at week 40.
Dose adjustment (every 8 weeks). At week 28, patients who were non-responders discontinued treatment and patients who were partial responders were adjusted to every-8-week dosing. PASI 75 responders at week 28 who became partial responders or non-responders at week 40 were adjusted to every-8-week dosing. All patients were followed for at least 52 weeks following first administration of study treatment.
PHOENIX 2. PHOENIX 2 evaluated the safety and efficacy of ustekinumab versus placebo in 1230 patients with plaque psoriasis. Patients randomised to ustekinumab received 45 mg or 90 mg doses at weeks 0 and 4 followed by an additional dose at week 16. Patients randomised to receive placebo at weeks 0 and 4 crossed over to receive ustekinumab (either 45 mg or 90 mg) at weeks 12 and 16. Patients were followed for 28 weeks.
Baseline disease characteristics: PHOENIX 1 and 2. Baseline disease characteristics across PHOENIX 1 and 2 were similar (Table 7).
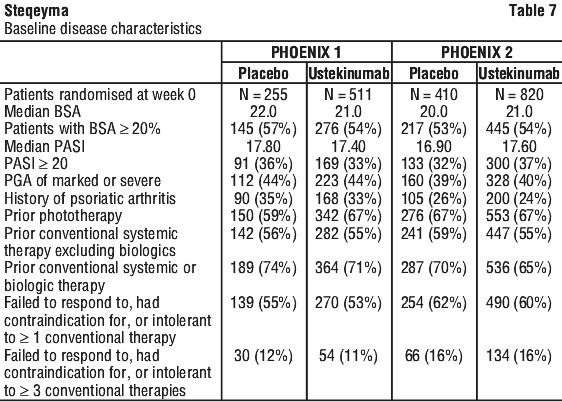
All 3 components of the PASI (plaque thickness/induration, erythema, and scaling) contributed comparably to the improvement in PASI.
The efficacy of ustekinumab was significantly superior (p < 0.001) to placebo across all subgroups defined by baseline demographics, clinical disease characteristics (including patients with a history of psoriatic arthritis) and prior medication usage. While pharmacokinetic modelling suggested a trend towards higher CL/F in patients with diabetes, a consistent effect on efficacy was not observed.
Other efficacy measures at week 12. In both PHOENIX 1 and PHOENIX 2, compared with placebo, significantly greater proportions of patients randomised to 45 mg or 90 mg ustekinumab achieved a cleared or minimal PGA score, and significantly greater proportions of patients randomised to 45 mg or 90 mg ustekinumab were PASI 90 and PASI 50 responders at Week 12 (Table 8). In the PHOENIX 1 study, 60% and 62% of the patients treated with 45 mg and 90 mg ustekinumab, respectively, achieved PGA scores of cleared or minimal compared with 4% of placebo-treated patients. In PHOENIX 2, 68% and 73% of patients receiving 45 mg or 90 mg ustekinumab, respectively, had cleared or minimal PGA scores compared with 5% of the placebo patients. In PHOENIX 1, PASI 90 was achieved by 42% and 37% of the patients treated with 45 mg and 90 mg ustekinumab, respectively, compared with 2% of placebo- treated patients. In PHOENIX 2, the percentage of patients achieving PASI 90 was 42% in the 45 mg ustekinumab group, 51% in the 90 mg ustekinumab group and 1% in the placebo group. The percentage of patients achieving PASI 50 in PHOENIX 1 was 84% and 86% in the 45 mg and 90 mg ustekinumab groups, respectively, compared with 10% in the placebo group. Similarly, 84% of patients treated with 45 mg ustekinumab, 89% of patients treated with 90 mg ustekinumab and 10% of patients treated with placebo reached PASI 50 in PHOENIX 2 (Table 8).
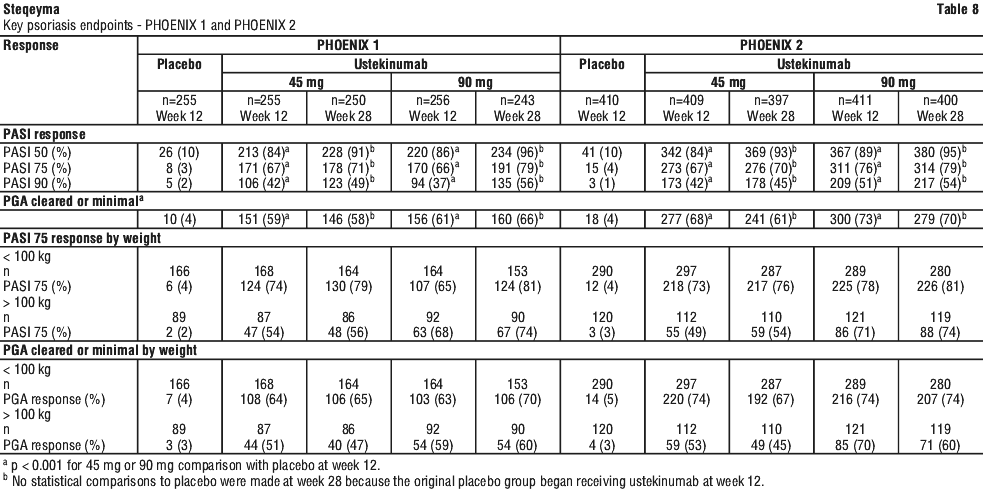
In pre-specified analyses of efficacy by body weight in PHOENIX 1 and PHOENIX 2, no consistent pattern of dose response was seen in patients ≤ 100 kg. In patients who weighed > 100 kg, higher PASI 75 response rates were seen with 90 mg dosing compared with 45 mg dosing, and a higher proportion of patients receiving 90 mg dosing had PGA scores of cleared or minimal compared with patients receiving 45 mg dosing (Table 8). Figure 1 shows PASI 75 response over time in PHOENIX 1 and 2.
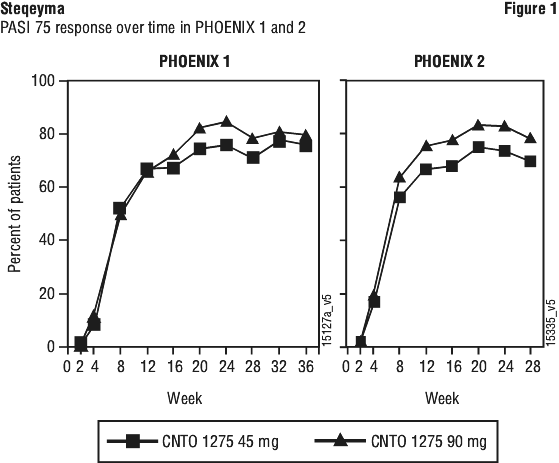
Efficacy of retreatment. In PHOENIX 1, after withdrawal from therapy, patients re-initiated their original ustekinumab treatment regimen after loss of ≥ 50% of PASI improvement. Retreatment with ustekinumab resulted in 76% of evaluated patients regaining PASI 75 response within 8 weeks after reinitiating therapy.
Dosing interval adjustment. In PHOENIX 1, week 28 and week 40 partial responders and week 40 non-responders were adjusted from every 12-week to every 8-week dosing. Approximately 40%-50% of week 28 partial responders to every 12-week dosing achieved PASI 75 response after adjustment to every 8-week dosing and this proportion of PASI 75 responders was maintained through week-52. A similar proportion of patients who were PASI 75 responders at week 28 and subsequently became partial responders or non-responders at week-40 achieved PASI 75 response following a dosing interval adjustment to every 8 weeks.
Quality of life. In PHOENIX 1 and 2, the mean baseline DLQI scores ranged from 11 to 12. In PHOENIX 1, the mean baseline SF-36 Physical Component ranged from 47-49 and the mean baseline SF-36 Mental Component was approximately 50. Quality of life improved significantly in patients randomised to 45 mg or 90 mg ustekinumab compared with patients randomised to placebo as evaluated by DLQI in PHOENIX 1 and 2 and SF-36 in PHOENIX 1. Quality of life improvements were significant as early as 2 weeks in patients treated with ustekinumab (p < 0.001) and these improvements were maintained over time with continued dosing.
In PHOENIX 1, 65% and 71% of patients treated with 45 mg and 90 mg of ustekinumab, respectively, showed a clinically meaningful reduction (5 or more points) in DLQI from baseline at week 12 compared to 18% in placebo group (p < 0.001 for both groups compared with placebo). Furthermore, 33% and 34% of patients treated with 45 mg and 90 mg of ustekinumab, respectively, showed a DLQI score of 0 compared to 1% in the placebo group (p < 0.001 for both groups compared with placebo), indicating no impairment in QOL from disease or treatment in these patients. In PHOENIX 2, 72% and 77% of patients treated with 45 mg and 90 mg of ustekinumab, respectively, showed a clinically meaningful reduction (5 or more points) in DLQI from baseline at week 12 compared to 21% in placebo group (p < 0.001 for both groups compared with placebo). In addition, 37% and 39% of patients treated with 45 mg and 90 mg of ustekinumab, respectively, showed a DLQI score of 0 compared to 1% in the placebo group (p < 0.001 for both groups compared with placebo).
In PHOENIX 1, the median baseline NAPSI score for nail psoriasis was 4.0 and the median number of fingernails involved with psoriasis was 8.0. Nail psoriasis improved significantly in patients randomised to 45 mg or 90 mg ustekinumab compared with patients randomised to placebo when measured by the NAPSI score (p ≤ 0.001). Improvements in physical and mental component summary scores of the SF-36 and in the Itch Visual Analogue Scale (VAS) were also significant in each ustekinumab treatment group compared with placebo (p < 0.001). In PHOENIX 2, the Hospital Anxiety and Depression Scale (HADS) and Work Limitations Questionnaire (WLQ) were also significantly improved in each ustekinumab treatment group compared with placebo (p < 0.001).
ACCEPT. A multicentre, randomised, single-blind, active-controlled study (ACCEPT) compared the safety and efficacy of ustekinumab and etanercept in patients 18 years of age and older with chronic (> 6 months) plaque psoriasis who had a minimum BSA involvement of 10%, PASI score ≥ 12, Physician Global Assessment (PGA) score ≥ 3, who were candidates for phototherapy or systemic therapy, and who had had an inadequate response to, intolerance to, or contraindication to ciclosporin, methotrexate, or PUVA therapy. A total of 903 patients were enrolled in the study.
The ACCEPT trial compared the efficacy of ustekinumab to etanercept and evaluated the safety of ustekinumab and etanercept in moderate to severe psoriasis patients. The active-controlled portion of the study was from week 0 to week 12, during which patients were randomised to receive etanercept (50 mg twice a week) ustekinumab 45 mg at weeks 0 and 4, or ustekinumab 90 mg at weeks 0 and 4. This trial was powered to test the superiority of each ustekinumab dose to etanercept on the primary endpoint of the proportion of patients who achieved a PASI 75 at week 12.
Significantly greater proportions of subjects treated with ustekinumab 45 mg (67%; p = 0.012) or 90 mg (74%; p < 0.001) were PASI 75 responders at week 12 compared with the etanercept group (56.8%). PASI 90 response was observed in 36% and 45% of patients in the ustekinumab 45 mg and 90 mg groups, respectively, compared with 23% of patients receiving etanercept (p < 0.001 for each comparison versus etanercept). PASI 100 response was observed in 12% and 21% of patients in the ustekinumab 45 mg and 90 mg groups, respectively, compared to 6% of patients receiving etanercept. In addition, a greater proportion of patients in the ustekinumab 45 mg and 90 mg treatment groups achieved a PGA score of "cleared" or "minimal" (65% and 71%, respectively) compared with patients in the etanercept treatment group (49%) (p < 0.001 for each comparison versus etanercept).
In pre-specified analyses of efficacy by body weight in ACCEPT, minimal dose response to ustekinumab was evident in patients ≤ 100 kg. In patients who weighed > 100 kg, higher PASI 75 response rates were seen with 90 mg dosing compared with 45 mg dosing, and a higher proportion of patients receiving 90 mg dosing had PGA scores of cleared or minimal compared with patients receiving 45 mg dosing (Table 9).
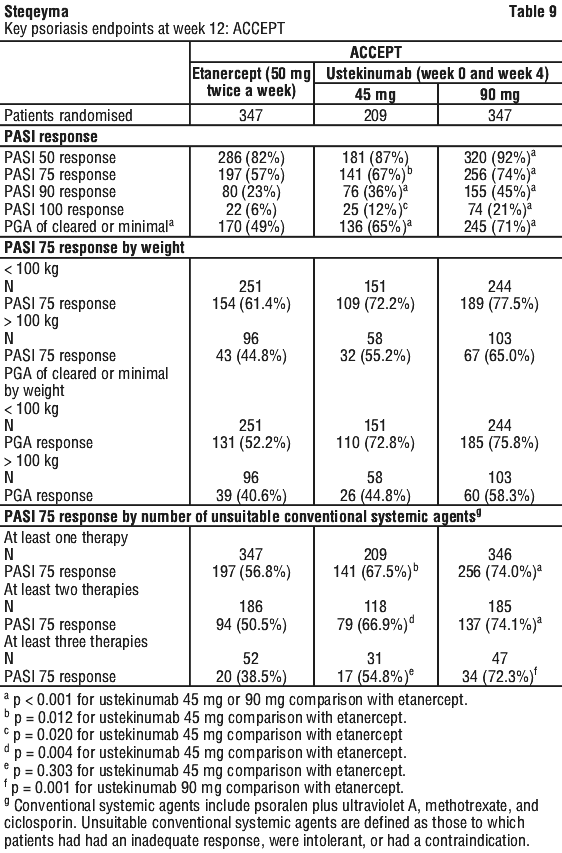
Patients with PASI ≥ 12, PGA ≥ 3 and BSA involvement of at least 10%, who were candidates for systemic or phototherapy, were eligible for the study. Approximately 60% of the patients had prior exposure to conventional systemic therapy or phototherapy. Approximately 11% of the patients had prior exposure to biologics.
The primary endpoint was the proportion of patients who achieved a PGA score of cleared (0) or minimal (1) at week 12. Secondary endpoints included PASI 75, PASI 90, change from baseline in Children's Dermatology Life Quality Index (CDLQI), and change from baseline in the total score of PedsQL (Paediatric Quality of Life Inventory) at week 12. At week 12, subjects treated with ustekinumab showed significantly greater improvement in their psoriasis and health related quality of life compared with placebo (Table 10).
All patients were followed for efficacy for up to 52 weeks following first administration of study agent. The proportion of patients with a PGA score of cleared (0) or minimal (1) and the proportion achieving PASI 75 showed separation between the ustekinumab treated group and placebo at the first post-baseline visit at week 4, reaching a maximum by week 12. Improvements in PGA, PASI, CDLQI and PedsQL were maintained through week 52 (Table 10).
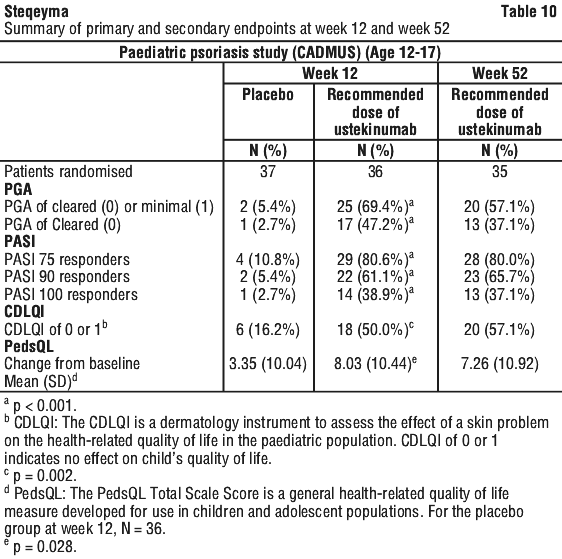
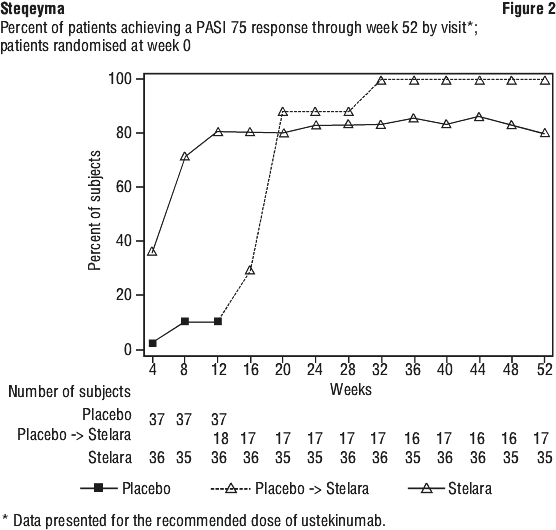
Plaque psoriasis (children - 6 to 11 years of age). The efficacy of ustekinumab was studied in 44 paediatric patients 6 to 11 years of age with moderate to severe plaque psoriasis in an open label, single-arm, multicentre, Phase 3 study (CADMUS Jr.). Patients were treated with the recommended dose of ustekinumab (n=44) (see Section 4.2 Dose and Method of Administration) by subcutaneous injection at weeks 0 and 4 followed by every 12 week (q12w) dosing.
Patients with PASI ≥ 12, PGA ≥ 3 and BSA involvement of at least 10%, who were candidates for systemic therapy or phototherapy, were eligible for the study. Approximately 43% of the patients had prior exposure to conventional systemic therapy or phototherapy. Approximately 5% of the patients had prior exposure to biologics.
The primary endpoint was the proportion of patients who achieved a PGA score of cleared (0) or minimal (1) at week 12. Secondary endpoints included PASI 75, PASI 90, and change from baseline in Children's Dermatology Life Quality Index (CDLQI) at week 12. At week 12, patients treated with ustekinumab showed clinically meaningful improvements in their psoriasis and health related quality of life (Table 10). All patients were followed for efficacy for up to 52 weeks following first administration of study agent. The proportion of patients with a PGA score of cleared (0) or minimal (1) at week 12 was 77.3%. Efficacy (defined as PGA 0 or 1) was observed as early as the first post-baseline visit at week 4 and the proportion of subjects who achieved a PGA score of 0 or 1 increased through week 16 and then remained relatively stable through week 52. Improvements in PGA, PASI, and CDLQI were maintained through week 52 (Table 11).
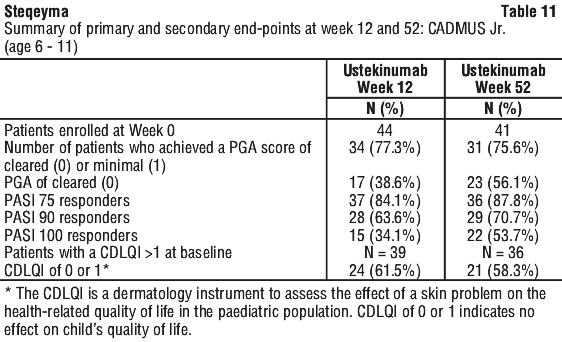
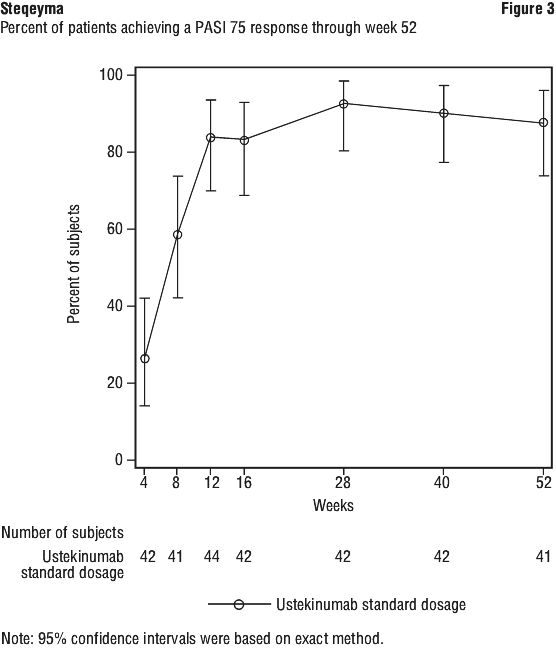
These studies included 927 (PSUMMIT I, n=615; PSUMMIT II, n=312) adult patients (≥ 18 years) who had active psoriatic arthritis (≥ 5 swollen joints and ≥ 5 tender joints, despite disease modifying antirheumatic (DMARD) and/or nonsteroidal anti-inflammatory (NSAID) therapy). Methotrexate use was allowed during the studies but was not mandatory. Approximately 50% of patients continued on stable doses of MTX (≤ 25 mg/week). In PSUMMIT I and PSUMMIT II, 80% and 86% of the patients, respectively, had been previously treated with DMARDs.
In PSUMMIT I, patients who had been previously treated with anti-TNFα therapy, prior to the first study dose, were excluded. In PSUMMIT II, the majority of patients (58%, n=180) had been previously treated with one or more anti-TNFα agent(s) for at least 8 weeks (14 weeks with infliximab) or had discontinued anti-TNFα for intolerance at any time. Among the patients who had been previously treated with an anti-TNFα agent, over 70% had discontinued their anti-TNFα treatment for lack of efficacy or intolerance.
Patients with each subtype of psoriatic arthritis were enrolled, including polyarticular arthritis with no evidence of rheumatic nodules (39%, n=362), spondylitis with peripheral arthritis (28%, n=255), asymmetrical peripheral arthritis (21%, n=193), distal interphalangeal (DIP) arthritis (12%, n=112) and arthritis mutilans (0.5%, n=5). Over 70% and 40% of the patients in both studies had enthesitis and dactylitis at baseline, respectively.
In both studies, a significantly greater proportion of patients achieved ACR 20 and ACR 50 responses at week 24 in the ustekinumab 45 mg and 90 mg groups compared to placebo (see Table 11). In PSUMMIT I, a significantly greater proportion of patients and in PSUMMIT II a numerically greater proportion of patients (p=NS) achieved ACR 70 responses in the ustekinumab 45 mg and 90 mg groups compared to placebo (see Table 11).
In both studies, the proportion of patients achieving a modified PsA response criteria (PsARC) or a Disease Activity Score 28 using C-reactive protein (DAS28-CRP) response was significantly greater in the ustekinumab 45 mg and 90 mg groups compared to placebo. In PSUMMIT I the proportion of patients achieving DAS28-CRP remission was significantly greater in the ustekinumab 45 mg and 90 mg groups compared to placebo. In PSUMMIT II, the proportion of patients who achieved DAS28- CRP remission was significantly greater in the ustekinumab 90 mg group compared to placebo (see Table 12). DAS28-CRP and PsARC responses were maintained through week 52.
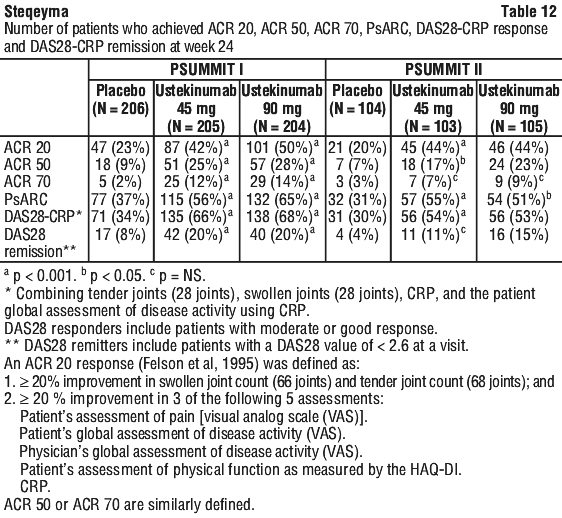
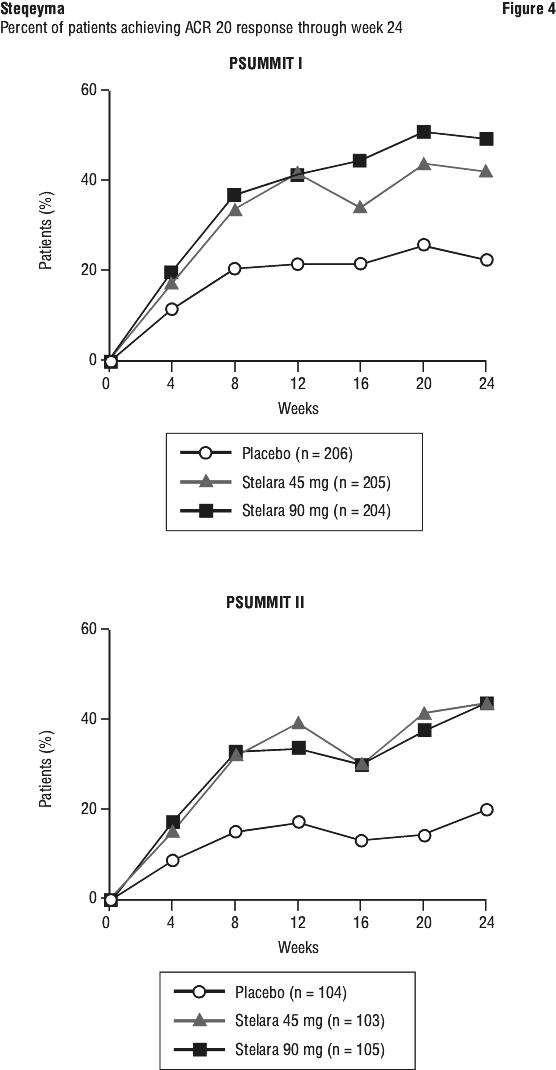

In PSUMMIT II, of 103 subjects randomised to ustekinumab 45 mg, 68 continued the same dose and were available for evaluation at week 52. Among those, ACR 20, 50 and 70 responses were achieved by 41 (60.3%), 23 (33.8%) and 11 (16.2%) subjects respectively. Of 105 subjects randomised to ustekinumab 90 mg, 83 were available for evaluation at week 52. Among those, ACR 20, 50 and 70 responses were achieved by 49 (59%), 26 (31.3%) and 17 (20.5%) subjects, respectively.
Additionally, within each weight group, (≤ 100 kg and > 100 kg), ACR 20, ACR 50 and ACR 70 responses were consistently higher in the ustekinumab 45 mg and 90 mg groups than in the placebo group (see Table 14).
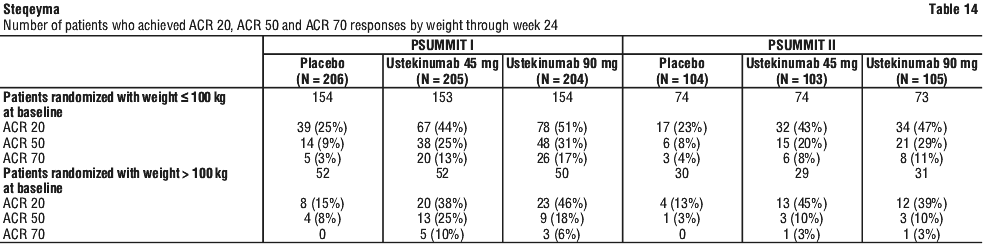

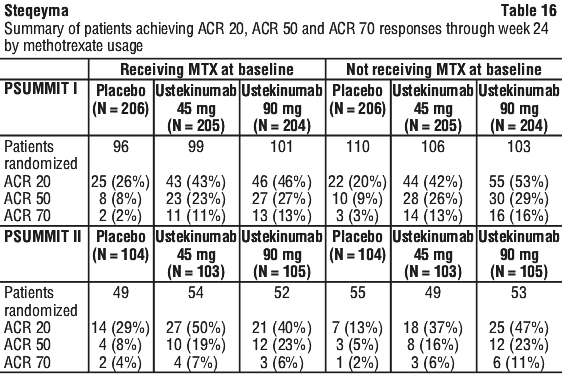
Among patients previously treated with anti-TNFα agents, a significantly greater proportion of ustekinumab-treated patients achieved an ACR 20 response at week 24 compared to placebo (see Table 17). ACR 20, 50 and 70 responses were generally maintained through week 52.
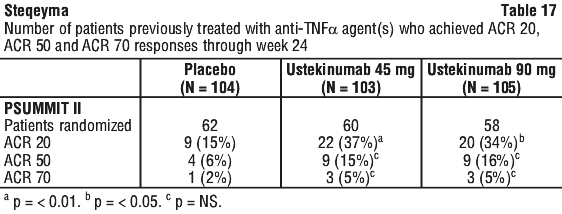
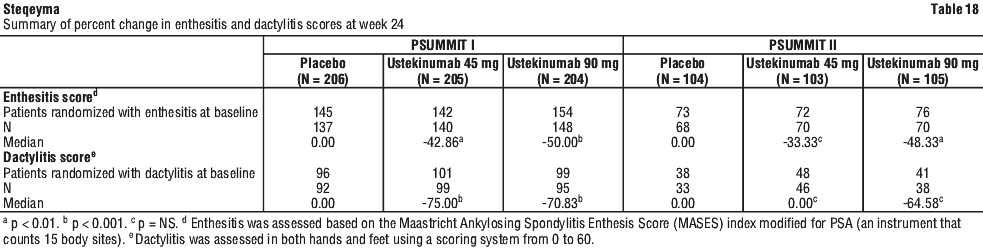
The proportion of patients who achieved both a PASI 75 response and an ACR 20 response was evaluated for those patients with ≥ 3% BSA psoriasis skin involvement at baseline. A significantly higher proportion of patients achieved the combined response in the ustekinumab 45 mg and 90 mg groups compared with the placebo group at week 24 (see Table 19). In both studies, the proportion of patients achieving both a PASI 75 response and an ACR20 response was maintained through week 52 (PSUMMIT I, ustekinumab 45 mg-44.8% and 90 mg-44.3%; PSUMMIT II, ustekinumab 45 mg-36.8% and 90 mg-43.1%).
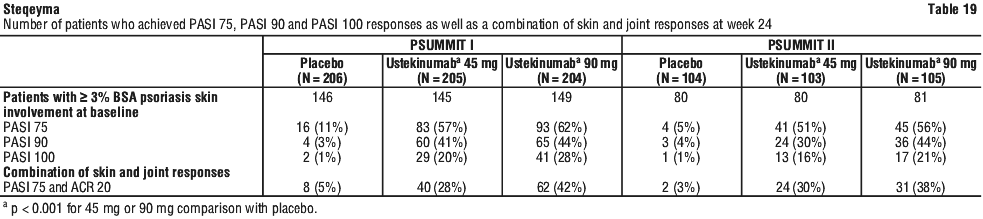
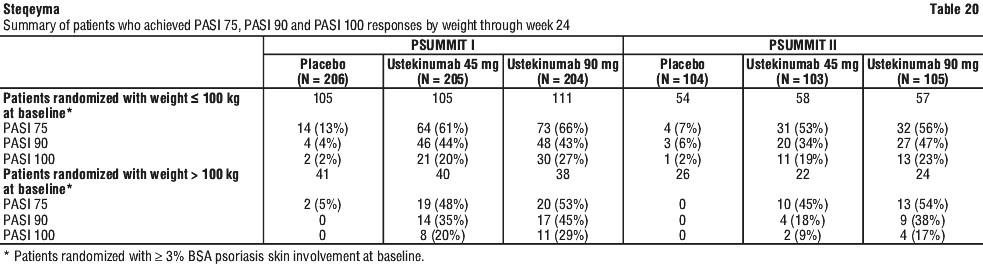
Prior anti-TNFα therapy. In PSUMMIT II, the proportion of patients who achieved a PASI 75 response at week 24 was significantly greater in ustekinumab 45 mg and 90 mg groups compared with placebo in patients previously treated with an anti-TNFα agent.
Radiographic response. Structural damage in both hands and feet was assessed by readers unaware of treatment group and order of visits and expressed as change in total van der Heijde-Sharp score (vdH-S score), modified for PsA by addition of hand distal interphalangeal (DIP) joints, compared to baseline. A pre-specified integrated analysis combining data from 927 subjects in both PSUMMIT I and II was performed. At week 24, based on this integrated analysis, the ustkinumab 45 mg or 90 mg treatment significantly inhibited progression of structural damage, when compared to placebo (see Table 20). Beyond week 24, ustekinumab treatment continued to inhibit the progression of structural damage through week 52. The mean change from week 24 to 52 in total modified vdH-S score (0.18 and 0.26 in the ustekinumab 45 mg and 90 mg groups respectively) was less than the mean change from week 0 to 24 (see Table 21).
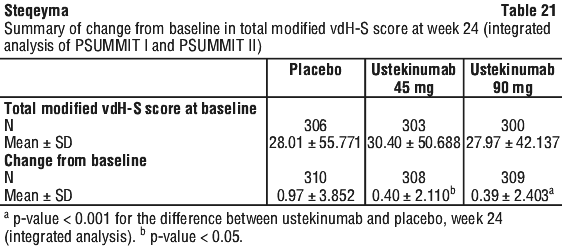
The effect of ustekinumab on progression of structural damage in patients with prior anti-TNFα experience has not been established.
Physical function and health-related quality of life. In PSUMMIT I and PSUMMIT II, physical function and health-related quality of life were assessed using the Disability Index of the Health Assessment Questionnaire (HAQ-DI), Dermatology Life Quality Index (DLQI) and the SF-36 health survey.
Patients treated with ustekinumab showed significant improvement in physical function as assessed by the HAQ-DI at week 24. The proportion of patients achieving a clinically meaningful ≥ 0.3 improvement in HAQ-DI score from baseline at week 24 was also significantly greater in the ustekinumab groups when compared with placebo (see Table 22). Improvement was observed at the first assessment (week 4), reached maximum at week 12 and was maintained through week 24. Improvement in HAQ-DI score from baseline was maintained at week 52.
In both studies, the improvement in HAQ-DI at week 24 was consistently greater in the ustekinumab 45 mg and 90 mg groups compared with placebo regardless of concomitant MTX use.
In PSUMMIT II, the improvement in HAQ-DI at week 24 was significantly greater in the ustekinumab 45 mg and 90 mg groups compared with placebo in patients previously treated with anti-TNFα agents.
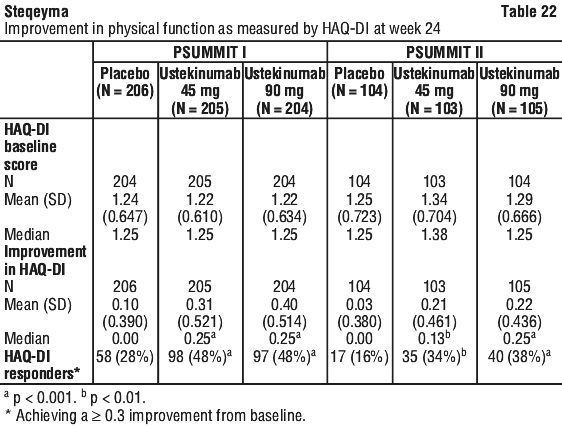
In PSUMMIT II, of 103 subjects randomised to ustekinumab 45 mg, 68 continued the same dose and were available for evaluation at week 52. Among those, the HAQ-DI response was achieved by 29 (42.6%) subjects. Of 105 subjects randomised to ustekinumab 90 mg, 83 were available for evaluation at week 52. Among those, HAQ-DI response was achieved by 44 (53%) subjects.
The DLQI was assessed by comparing the change in DLQI scores from baseline for those patients with ≥ 3% BSA at baseline. In both studies at week 24, there was a significant improvement from baseline in DLQI scores in both the ustekinumab 45 mg and 90 mg groups as compared with placebo (see Table 23) and the improvement was maintained at week 52.
In both PSUMMIT I and PSUMMIT II, at week 24, the change from baseline in the SF-36 physical component summary (PCS) scores was significantly greater in the ustekinumab 45 mg and 90 mg groups compared with the placebo group. In both studies, the change from baseline in the SF-36 mental component summary (MCS) scores at week 24 was greater in both ustekinumab groups compared with the placebo group (p < 0.001 for PSUMMIT I - 90 mg group, p=NS for other groups) (see Table 23). In both studies, the change from baseline in the SF-36 PCS and MCS scores was maintained at week 52.
In PSUMMIT II, a significant change from baseline in Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F) scores was observed at week 24 in the ustekinumab 45 mg and 90 mg groups compared with the placebo group (median improvement, all 3.0 vs 0.0; p < 0.007). Similarly, the percentage of patients with clinically significant improvement in fatigue from baseline (4 points in FACIT-F) was significantly greater in the ustekinumab 45 mg (49% [p < 0.001]) and 90 mg groups (49% [p < 0.001]) compared with the placebo group (25.8%). The change from baseline in the FACIT- F scores was maintained at week 52.
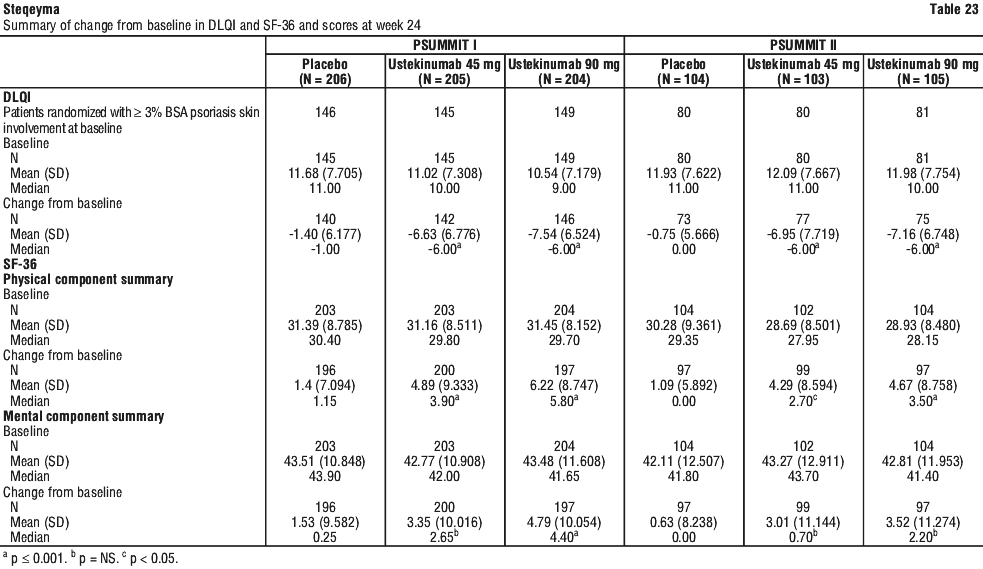
Induction of clinical response and remission. UNITI-1 and UNITI-2 studies included 1409 (UNITI-1 n=769; UNITI-2 n=640) patients. In both studies, patients were permitted to concomitantly receive oral 5-ASA compounds, immunomodulators, corticosteroids, and/or antibiotics. Patients were randomised to receive a single IV administration of ustekinumab, designed as a tiered dose based on patient body weight (Table 3) or placebo at week 0. The primary endpoint was clinical response (defined as a reduction in CDAI score of ≥ 100 points or CDAI score < 150) at week 6. Secondary endpoints included clinical remission at week 8, clinical response at week 8, 70-point response at week 3, and 70-point response at week 6. Efficacy data were collected and analysed through week 8 for both studies.
In UNITI-1, patients had failed or were intolerant to prior anti-TNFα therapy. At baseline, approximately 46% (n=340) patients were receiving corticosteroids (including budesonide) and 31.4% of patients were receiving immunomodulators. Approximately 48% had failed 1 prior anti- TNFα therapy and 52% had failed 2 or 3 prior anti-TNFα therapies (40.8% and 10.4%, respectively). In this study, 29.1% patients had an inadequate initial response (primary non-responders), 69.4% responded but subsequently lost response (secondary non-responders), and 36.4% were intolerant to anti-TNF α therapies.
Patients in UNITI-2 had failed at least one conventional therapy (corticosteroids or immunomodulators) and were either anti-TNFα naïve (68.6%) or had previously received but not failed anti-TNFα therapy (31.4%). At baseline, approximately 40% patients were receiving corticosteroids (including budesonide) and 35% patients were receiving immunomodulators.
In these induction studies, efficacy was higher and better sustained in the tiered dose (based on weight ranges) group compared to the 130 mg dose group. In both UNITI-1 and UNITI-2, a significantly greater proportion of patients were in clinical response and remission in the group treated with ustekinumab, compared to placebo (Table 24, Figure 5). Clinical response and remission were significant as early as week 3 in ustekinumab treated patients and continued to improve through week 8 (Figure 5).
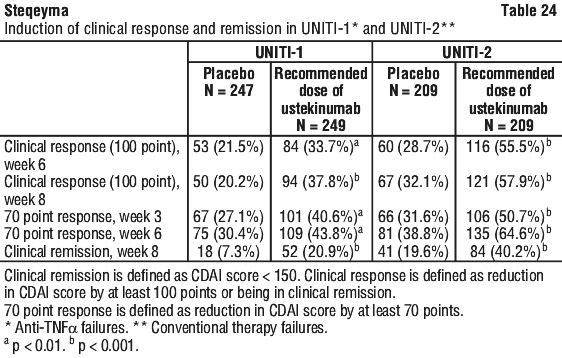
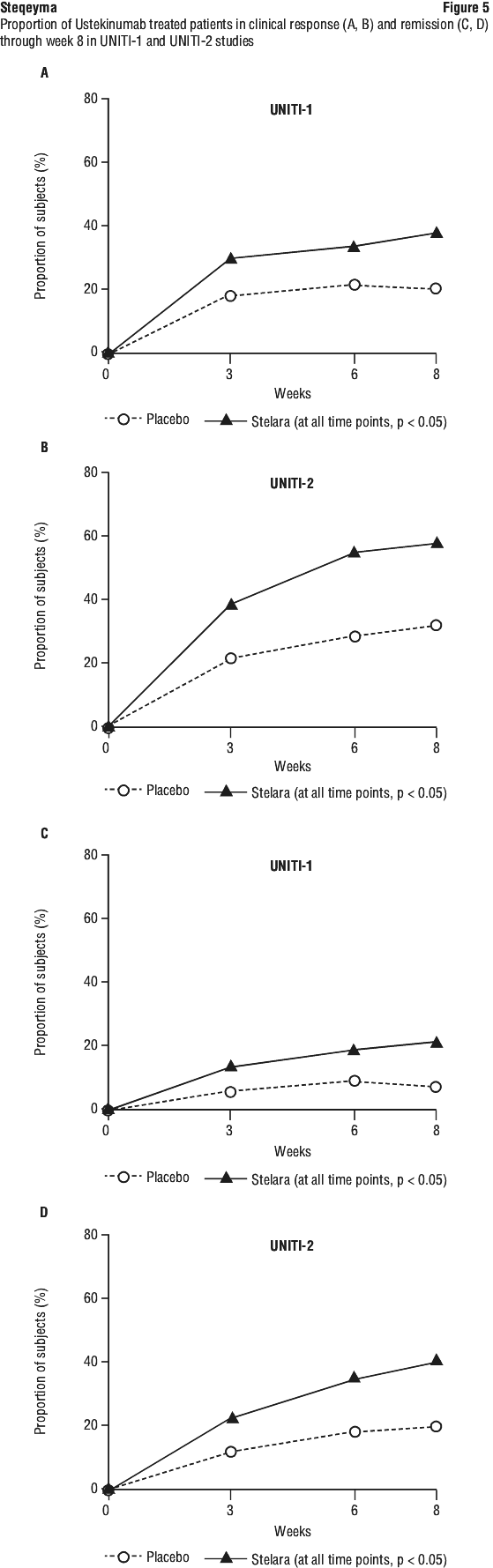
Concomitant doses of oral 5-ASA compounds, immunomodulators corticosteroids and antibiotics were permitted. Corticosteroids were tapered at the start of the maintenance trial. The primary endpoint was clinical remission (CDAI < 150) at week 44. Secondary endpoints assessed at week 44 included clinical response, clinical remission among ustekinumab treated patients in clinical remission after induction, corticosteroid-free remission, and clinical remission in the subset of patients who were refractory or intolerant to anti-TNFα treatment.
Significantly higher proportions of patients maintained clinical remission and response in the ustekinumab treated groups as compared to placebo at week 44 (Table 24, Figure 6). A higher proportion of ustekinumab treated patients compared to placebo achieved sustained clinical remission (clinical remission at week 36, 40 and 44). Clinical remission was achieved in patients who had failed conventional therapy (anti-TNFα naïve) and in patients who had prior treatment experience with an anti-TNFα. A higher rate of clinical remission was observed in the anti-TNFα naïve patients compared to the anti-TNFα refractory/intolerant patients, but the overall treatment effect was consistent in both anti-TNFα refractory/intolerant patients and anti-TNFα naïve patients (Table 25).
Dosing in patients with a lower inflammatory burden. In patients with a lower inflammatory burden as reflected by CRP ≤ 10 mg/L at initiation of induction or initiation of maintenance therapy, the efficacy of the every 12 week dosing regimen was similar to that of the every 8 week dosing regimen.
Dosing frequency adjustment. In IM-UNITI, 29 of 129 patients (22%) did not maintain response to ustekinumab when treated every 12 weeks and were allowed to increase the frequency of dosing and receive ustekinumab every 8 weeks. In these patients, clinical remission was achieved in 41.4% of patients 16 weeks after dosing frequency adjustment.
Resumption of treatment. Of 131 patients that responded to ustekinumab induction and who were randomised to the placebo group at the start of the maintenance study, 51 subsequently lost response and received 90 mg ustekinumab subcutaneously every 8 weeks. Of these 51 patients, 70.6% achieved clinical response and 39.2% achieved clinical remission 16 weeks after receiving the first subcutaneous dose of ustekinumab.
Long-term maintenance. In IM-UNITI, patients who completed the study through week 44 were eligible to continue treatment in a study extension. Among patients who entered the study extension, clinical remission and response were generally maintained through week 252. Results were consistent between patients who failed TNF-therapies versus those who did not.
No new safety concerns were identified in this study extension with up to 5 years of treatment in patients with Crohn's disease.
Corticosteroid use in maintenance. In patients that were in clinical response to ustekinumab induction therapy, a greater proportion of patients in the ustekinumab treated group were in remission and corticosteroid-free compared to the placebo group after 44 weeks of maintenance treatment (Table 24). In addition, a higher proportion of patients were in clinical response and not receiving corticosteroids in the ustekinumab treated group compared to placebo.
Endoscopic healing of the mucosa. Endoscopic healing of the mucosa was evaluated in 252 patients with baseline endoscopic disease activity in a substudy. At Week 8, after a single IV induction dose, reduction in mucosal inflammation, as measured by the Simplified Endoscopic Activity Score for Crohn's Disease (SES-CD), was greater in patients treated with ustekinumab (n=83) compared with patients treated with placebo (n=97) (-2.8 vs -0.7, p=0.012). Similar reductions in histologic inflammation were also observed.
Fistula response. In patients with draining fistulas at baseline (8.8%), a numerically greater proportion of ustekinumab treated patients achieved a fistula response (defined as ≥ 50% reduction from baseline of the induction study in the number of draining fistulas) compared with placebo over 44 weeks (p=NS). The proportion of patients in fistula response at Week 44 was 45.5% (5/11) for placebo group, 71.4% (5/7) for ustekinumab 90 mg every 12 week dosing group, and 87.5% (7/8) for ustekinumab 90 mg every 8 week dosing group.
Health-related quality of life measures. Improvement in general and disease specific health-related quality of life was assessed using the SF-36 and Inflammatory Bowel Disease Questionnaire (IBDQ) respectively.
SF-36. A higher proportion of patients treated with ustekinumab showed clinically meaningful improvements in SF-36 Physical Component Summary (PCS) and Mental Component Summary (MCS) scores, and these improvements were significantly greater at week 8 compared with the placebo group in UNITI-1 (MCS) and UNITI-2 (PCS, MCS and all subscores). These improvements in the PCS and MCS scores were maintained in ustekinumab treated patients in the IM-UNITI maintenance study through Week 44.
IBDQ. At week 8 in UNITI-1 and UNITI-2, significant improvement from baseline in the inflammatory bowel disease questionnaire (IBDQ) total score and all subscales, was observed in the patients treated with ustekinumab compared to placebo. In both studies, a higher proportion of patients with clinically meaningful improvement in IBDQ total scores were observed in patients treated with ustekinumab compared to placebo. These improvements in the IBDQ total scores were maintained in ustekinumab treated patients in the IM-UNITI maintenance study through week 44.
Long-term maintenance of health-related quality of life measures. The majority of subjects maintained a 16-point improvement in IBDQ score and a 5-point improvement in SF-36 through week 252.
Ulcerative colitis. The safety and efficacy of ustekinumab was assessed in two randomised, double-blind, placebo controlled, multicenter studies in adult patients with moderately to severely active ulcerative colitis (Mayo score 6 to 12; Endoscopy subscore ≥ 2 based on central review of the endoscopy). The clinical development program consisted of one IV induction study (referred to as UNIFI-induction) with treatment of up to 16 weeks followed by a 44-week subcutaneous randomized withdrawal maintenance study (referred to as UNIFI-maintenance) representing at least 52 weeks of therapy.
Efficacy results presented for UNIFI-induction and UNIFI-maintenance were based on central review of endoscopies.
UNIFI-induction included 961 patients. The primary endpoint for the induction study was the proportion of patients in clinical remission (defined as a Mayo score ≤ 2 points, with no individual subscore > 1) at Week 8. Patients were randomized to receive a single IV administration of either the recommended tiered dose of approximately 6 mg/kg (see Table 3; Initial IV dosing of ustekinumaba), a fixed dose of 130 mg ustekinumab, or placebo at week 0.
Concomitant use of oral corticosteroids, immunomodulators, and aminosalicylates were permitted and 90% of patients continued to receive at least one of these medications. Enrolled patients had to have failed conventional therapy (corticosteroids or immunomodulators) or at least one biologic (a TNFα antagonist and/or vedolizumab). 49% of patients had failed conventional therapy, but not a biologic (of which 94% where biological-naïve). 51% of patients had failed or were intolerant to a biologic. Approximately 50% of the patients had failed at least 1 prior anti-TNFα therapy (of which 48% were primary non-responders) and 17% had failed at least 1 anti-TNFα therapy and vedolizumab.
In UNIFI-induction a significantly greater proportion of patients were in clinical response and remission in the ustekinumab treated group compared to placebo (Table 26). As early as week 2, the earliest scheduled study visit, and at each visit thereafter, a higher proportion of ustekinumab patients had no rectal bleeding or achieved normal stool frequency (defined as a stool frequency subscore of 0 or 1) as compared with placebo patients. Significant differences in partial Mayo score and symptomatic remission were observed between ustekinumab and placebo as early as week 2. Efficacy was higher in the tiered dose group (6 mg/kg) compared to the 130 mg dose group in select endpoints, and tiered dosing is therefore the recommended IV induction dose.
Significantly greater proportions of patients were in clinical remission at week 44 and maintained clinical response through week 44 in both ustekinumab treated groups compared to the placebo group (see Table 27).
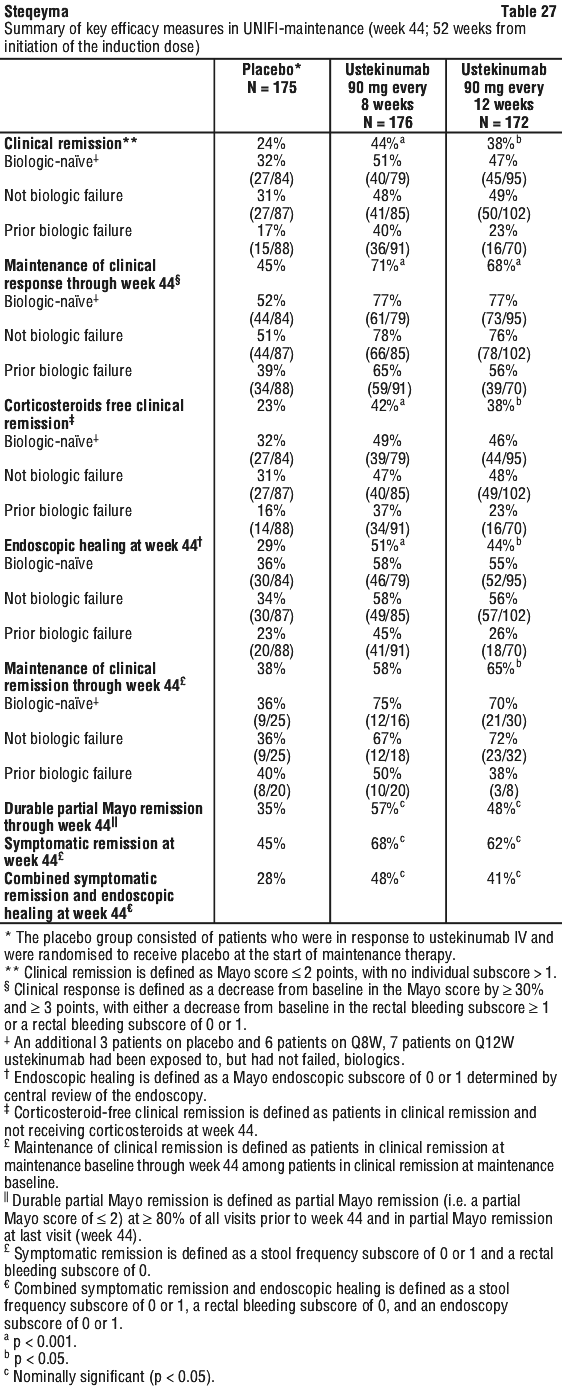
Delayed responders to ustekinumab induction. Ustekinumab treated patients who were not in response at week 8 of UNIFI-induction received an administration of 90 mg SC ustekinumab at week 8 (36% of patients). Of those patients, 9% of patients who were initially randomized to the recommended induction dose achieved clinical remission and 58% achieved clinical response at week 16. When combining the delayed responders with the initial responders, 80% of subjects randomized to the recommended induction dose in UNIFI-I achieved clinical response and 18% achieved clinical remission within 16 weeks after initiating treatment with ustekinumab.
Patients who were not in clinical response to ustekinumab induction at week 8 of the UNIFI-induction study but were in response at week 16 (157 patients) entered in the non-randomized portion of UNIFI-maintenance and continued to receive maintenance dosing every 8 weeks; among these patients, a majority (62%) maintained response and 30% achieved remission at week 44.
Long-term maintenance. In UNIFI (UCO3001), patients who completed the study through week 44 were eligible to continue treatment in a study extension. Among patients who entered the study extension and were treated with ustekinumab, the majority of patients who failed conventional therapy (but not a biologic therapy) and those who failed biologic therapy, including those who failed both anti-TNF and vedolizumab maintained a response at week 200. Of those patients who were assessed using the full Mayo score at week 200, mucosal healing and clinical remission were generally maintained.
No new safety concerns were identified in this study extension with up to 4 years of treatment in patients with ulcerative colitis.
Endoscopic normalisation. Normalization of endoscopic appearance of the mucosa was defined as a Mayo endoscopic subscore of 0 and was observed as early as week 8 of UNIFI-induction. At week 44 of UNIFI- maintenance, it was achieved in 24% and 29% of patients treated with ustekinumab every 12 or 8 weeks, respectively, as compared to 18% of patients in the placebo group.
Histologic and histo-endoscopic mucosal healing. Histologic healing (defined as neutrophil infiltration in < 5% of crypts, no crypt destruction, and no erosions, ulcerations, or granulation tissue) was assessed at week 8 of UNIFI-induction and week 44 of UNIFI-maintenance. At week 8, after a single IV induction dose, significantly greater proportions of patients in the recommended dose group achieved histologic healing (36%) compared with patients in the placebo group (22%). At week 44 maintenance of this effect was maintained with significantly more patients in histologic healing in the every 12 week (54%) and every 8 week (59%) ustekinumab groups as compared to placebo (33%).
A combined endpoint of histo-endoscopic mucosal healing defined as subjects having both mucosal healing and histologic healing was evaluated at week 8 of UNIFI-induction and week 44 of UNIFI-maintenance. Patients receiving ustekinumab at the recommended dose showed significant improvements on the histo-endoscopic mucosal healing endpoint at week 8 in the ustekinumab group (18%) as compared to the placebo group (9%). At week 44, maintenance of this effect was observed with significantly more patients in histo-endoscopic mucosal healing in the every 12 week (39%) and every 8 week (46%) ustekinumab groups as compared to placebo (24%).
Health-related quality of life. Health-related quality of life was assessed by Inflammatory Bowel Disease Questionnaire (IBDQ), SF-36 and EuroQoL-5D (EQ-5D) questionnaires. At Week 8 of UNIFI-induction, patients receiving ustekinumab showed significantly greater and clinically meaningful improvements on IBDQ total score, EQ-5D and EQ-5D VAS, and SF-36 Mental Component Summary Score and SF-36 Physical Component Summary Score when compared to placebo. These improvements were maintained in ustekinumab-treated patients in UNIFI-maintenance through week 44.
Patients receiving ustekinumab experienced significantly more improvements in work productivity as assessed by greater reductions in overall work impairment and in activity impairment as assessed by the WPAI-GH questionnaire than patients receiving placebo.
Long-term maintenance of health-related quality of life measures. Improvement in health-related quality of life as measured by IBDQ and SF-36 was generally maintained during the extension through week 200.
Hospitalisations and ulcerative colitis related surgeries. Through week 8 of UNIFI-induction, the proportions of subjects with ulcerative colitis disease related hospitalizations were significantly lower for subjects in the ustekinumab recommended dose group (1.6%, 5/322) compared with subjects in the placebo group (4.4%, 14/319) and no subjects underwent ulcerative colitis disease related surgeries in subjects receiving ustekinumab at the recommended induction dose compared to 0.6% (2/319) subjects in the placebo group.
Through week 44 of UNIFI-maintenance, a significantly lower number of ulcerative colitis disease related hospitalizations was observed in subjects in the combined ustekinumab group (2.0%, 7/348) as compared with subjects in the placebo group (5.7%, 10/175). A numerically lower number of subjects in the ustekinumab group (0.6%, 2/348) underwent ulcerative colitis disease related surgeries compared with subjects in the placebo group (1.7%, 3/175) through week 44.
5.2 Pharmacokinetic Properties
Absorption. The median time to reach the maximum serum concentration (tmax) was 8.5 days after a single 90 mg subcutaneous administration in healthy subjects. The median tmax values of ustekinumab following a single subcutaneous administration of either 45 mg or 90 mg in patients with psoriasis were comparable to that observed in healthy subjects.
The absolute bioavailability of ustekinumab following a single subcutaneous administration was estimated to be 57.2% in patients with psoriasis. Following the recommended IV induction dose, median peak serum ustekinumab concentration was 126.1 microgram/mL in patients with Crohn's disease, and 127.0 microgram/mL in patients with ulcerative colitis.
Distribution. Median volume of distribution during the terminal phase (Vz) following a single IV administration to patients with psoriasis, ranged from 57 to 83 mL/kg. In a population pharmacokinetic analysis of ustekinumab, the volume of distribution at steady-state was 4.62 L in patients with Crohn's disease and 4.44 L in patients with ulcerative colitis.
Metabolism. The exact metabolic pathway for ustekinumab is unknown.
Excretion. Median systemic clearance (CL) following a single IV administration to patients with psoriasis ranged from 1.99 to 2.34 mL/day/kg. Median half-life (t1/2) of ustekinumab was approximately 3 weeks in patients with ulcerative colitis, Crohn's disease, psoriasis and/or psoriatic arthritis, ranging from 15 to 32 days across all psoriasis and psoriatic arthritis studies. In a population pharmacokinetic analysis of ustekinumab, the clearance was 0.19 L/day while the half-life was approximately 19 days in patients with Crohn's disease and ulcerative colitis.
Dose linearity. The systemic exposure of ustekinumab (Cmax and AUC) increased in an approximately dose-proportional manner after a single IV administration at doses ranging from 0.09 mg/kg to 4.5 mg/kg or following a single subcutaneous administration at doses ranging from approximately 24 mg to 240 mg in patients with psoriasis.
Single dose vs. multiple doses. Serum concentration-time profiles of ustekinumab were generally predictable after single or multiple subcutaneous dose administrations. In patients with psoriasis, steady-state serum concentrations of ustekinumab were achieved by week 28 after initial subcutaneous doses at weeks 0 and 4, followed by doses every 12 weeks. The median steady-state trough concentration ranged from 0.21 microgram/mL to 0.26 microgram/mL (45 mg dose) and from 0.47 microgram/mL to 0.49 microgram/mL (90 mg dose).
Following the recommended IV induction dose, median peak serum ustekinumab concentration was 126.1 microgram/mL in patients with Crohn's disease and 127.0 microgram/mL in patient with ulcerative colitis. Starting at week 8, subcutaneous maintenance dosing of 90 mg ustekinumab was administered every 8 or 12 weeks. Steady state ustekinumab concentration was achieved by the start of the second maintenance dose. There was no apparent accumulation in serum ustekinumab concentration over time when given subcutaneously every 8 or 12 weeks.
Following subcutaneous maintenance dosing of 90 mg ustekinumab every 8 weeks, median steady- state trough concentrations ranged from 1.97 microgram/mL to 2.24 microgram/mL in patients with Crohn's disease and 2.69 microgram/mL to 3.09 microgram/mL in patients with ulcerative colitis. Following subcutaneous maintenance dosing of 90 mg ustekinumab every 12 weeks, median steady state trough concentrations ranged from 0.61 microgram/mL to 0.76 microgram/mL in patients with Crohn's disease and 0.92 microgram/mL to 1.19 microgram/mL in patients with ulcerative colitis. The steady-state trough ustekinumab levels resulting from 90 mg ustekinumab every 8 weeks were associated with higher clinical remission rates as compared to the steady-state trough levels following 90 mg every 12 weeks.
Dosing frequency adjustment. In patients with Crohn's disease and ulcerative colitis, based on observed data and population PK analyses, randomised subjects who lost response to treatment had lower serum ustekinumab concentrations over time compared with subjects who did not lose response. In Crohn's disease, dose adjustment from 90 mg every 12 weeks to 90 mg every 8 weeks was associated with an increase in trough serum ustekinumab concentrations and an accompanying increase in efficacy. In ulcerative colitis, population PK model based simulations demonstrated that adjusting dosing from 90 mg every 12 weeks to every 8 weeks would be expected to result in a 3-fold increase in steady-state trough ustekinumab concentrations. Additionally on the basis of clinical trial data in patients with ulcerative colitis, a positive exposure-response relationship was established between trough concentrations and clinical response, clinical remission, and mucosal healing.
Impact of weight on pharmacokinetics. Serum ustekinumab concentrations were affected by weight in patients with psoriasis and/or psoriatic arthritis. Within each dose (45 or 90 mg), patients of higher weight (> 100 kg) had lower median serum ustekinumab concentrations compared with those in patients of lower weight (≤ 100 kg). However, across doses, the median trough serum concentrations of ustekinumab in patients with higher weight (> 100 kg) in the 90 mg group were comparable to those in patients with lower weight (≤ 100 kg) in the 45 mg group.
Population pharmacokinetic analysis. In a population pharmacokinetic analysis using data from patients with psoriasis, the apparent clearance (CL/F) and apparent volume of distribution (V/F) were 0.465 L/d and 15.7 L, respectively, and the t1/2 was approximately 3 weeks. The CL/F of ustekinumab was not impacted by sex, age, or race. The CL/F was impacted by body weight, with a trend toward higher CL/F in patients with higher body weight. The median CL/F in patients with weight > 100 kg was approximately 55% higher compared with patients with weight < 100 kg. The median V/F in patients with weight > 100 kg was approximately 37% higher as compared with patients with weight < 100 kg. Similar results were obtained from a confirmatory population pharmacokinetic analysis using data from patients with psoriatic arthritis.
In the population pharmacokinetic analysis using data from patients with psoriasis, the effect of comorbidities (past and current history of diabetes, hypertension, and hyperlipidaemia) on pharmacokinetics of ustekinumab was evaluated. The pharmacokinetics of ustekinumab were impacted by the comorbidity of diabetes, with a trend towards higher CL/F in patients with diabetes. The mean CL/F in patients with diabetes was approximately 29% higher compared with patients without diabetes.
No specific drug-drug interaction studies have been conducted in healthy subjects or patients with psoriasis, psoriatic arthritis, Crohn's disease or ulcerative colitis.
In the population pharmacokinetic analyses, the effect of the most frequently used concomitant medications in patients with psoriasis (including paracetamol/acetaminophen, ibuprofen, acetylsalicylic acid, metformin, atorvastatin, naproxen, levothyroxine, hydrochlorothiazide, and influenza vaccine) on pharmacokinetics of ustekinumab was explored and none of the concomitant medications exerted significant impact. The pharmacokinetics of ustekinumab was not impacted by the prior use of MTX, ciclosporin, or other biological therapeutics for the treatment of psoriasis. The pharmacokinetics of ustekinumab was not impacted by concomitant use of NSAIDs or prior exposure to anti-TNFα agents in patients with psoriatic arthritis; or by the use of MTX, oral corticosteroids, 6- MP, AZA in patients with psoriatic arthritis or Crohn's disease, or by prior exposure to biologics (i.e. anti-TNFα agents and/or vedolizumab) in patients with ulcerative colitis.
No pharmacokinetic data are available in patients with renal insufficiency. No pharmacokinetic data are available in patients with impaired hepatic function.
No specific studies have been conducted in elderly patients. The population pharmacokinetic analysis indicated there were no apparent changes in CL/F and V/F estimates in patients > 65 years.
The pharmacokinetics of ustekinumab were not impacted by the use of tobacco or alcohol.
5.3 Preclinical Safety Data
Genotoxicity. Genotoxic potential has not been evaluated for ustekinumab.
Carcinogenicity. Ustekinumab has not been evaluated for carcinogenic potential, due to the lack of appropriate models for an antibody with no cross-reactivity to rodent IL-12/23 p40. Ustekinumab is a selective immunosuppressant agent. Immunosuppressive agents have the potential to increase the risk of malignancy (see Section 4.4 Special Warnings and Precautions for Use, Malignancies).
6 Pharmaceutical Particulars
6.1 List of Excipients
Vial (45mg), pre-filled syringe (45 mg, 90 mg) and pre-filled pen (45 mg, 90 mg). Each mL of Steqeyma solution for injection for subcutaneous administration contains: (see Table 28).


6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products.
Steqeyma 130 mg concentrate for solution for infusion should only be diluted with sodium chloride 9 mg/mL (0.9%) solution. Steqeyma 130 mg concentrate for solution for infusion should not be administered concomitantly in the same IV line with other medicinal products.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store at 2°C to 8°C. Refrigerate. Do not freeze.
Protect from light by storing in original carton until time of use. Do not shake.
To reduce microbiological hazard, use as soon as practicable after dilution. If storage is necessary, the diluted infusion solution may be stored for up to 48 hours at 2-8°C or eight hours at room temperature. Do not freeze. Discard any unused portion of the infusion solution.
Steqeyma pre-filled syringes, pre-filled pens and 45 mg/0.5 mL vial - room temperature storage. If needed, Steqeyma pre-filled syringes and pre-filled pens may be stored at room temperature up to a maximum of 25°C for a single period of up to 31 days within the shelf-life in the original carton protected from light. Once the syringe or pen has been stored at room temperature, it should not be returned to the refrigerator. Discard the syringe or pen if not used within 31 days at room temperature storage.
If needed, Steqeyma 45 mg/0.5 mL vials may be stored at room temperature up to a maximum of 30°C for a single period of up to 15 days in the original carton protected from light. Record the date when the vial is first removed from the refrigerator on the carton in the space provided. The new expiry date must not exceed the original expiry date printed on the carton. Once a vial has been stored at room temperature, it should not be returned to the refrigerator. Discard the vial if not used within 15 days at room temperature storage.
6.5 Nature and Contents of Container
For subcutaneous administration. Steqeyma is supplied as a sterile solution in a single-use (Type 1) glass vial. The vial is stoppered with a coated stopper.
Steqeyma is supplied as a single-use, sterile solution in a Type 1 glass pre-filled syringe with a fixed 27G, half-inch needle and needle cover. The syringe is fitted with a passive safety guard.
Steqeyma is also supplied as a pre-filled pen for adult use.
The solution is clear to slightly opalescent, colourless to pale yellow with a pH of approximately 5.7. Steqeyma does not contain preservatives.
Each pre-filled syringe, pre-filled pen and vial is for single use only and any unused medicinal product should be disposed of in accordance with local requirements.
Steqeyma for subcutaneous administration is available in two strengths: 45 mg of ustekinumab (rch) in 0.5 mL in vial, pre-filled syringe and pre-filled pen, or 90 mg of ustekinumab (rch) in 1.0 mL in pre-filled syringe and pre-filled pen.
Steqeyma is available in packs of:
1 single use vial (45 mg), 1 single use pre-filled syringe (45 mg or 90 mg) and 1 pre-filled pen (45 mg or 90 mg).
For IV infusion only. Steqeyma 130 mg vial is supplied as a sterile solution in a single-use (Type 1) glass vial. The vial is stoppered with a coated stopper.
The solution is clear to slightly opalescent, colourless to pale yellow with a pH of approximately 5.7. Steqeyma does not contain preservatives.
Steqeyma is available for IV infusion in one strength, 130 mg in 26 mL, and packaged as 1 single use vial. Product is for single use in one patient only. Discard any residue.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
Steqeyma (ustekinumab) is a human IgG1 kappa monoclonal antibody with an approximate molecular weight of 145,390 daltons. Steqeyma is produced by a recombinant cell line cultured by continuous perfusion and is purified by a series of steps that includes measures to inactivate and remove viruses.
7 Medicine Schedule (Poisons Standard)
S4 - Prescription Only Medicine.
Date of First Approval
02 September 2024
Date of Revision
06 January 2026
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.