Teglutik Oral Suspension
Brand Information
| Brand name | Teglutik Oral Suspension |
| Active ingredient | Riluzole |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Teglutik Oral Suspension.
Summary CMI
Teglutik® Oral Suspension
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I using Teglutik®?
Teglutik® contains the active ingredient riluzole. Teglutik® is used to treat people with amyotrophic lateral sclerosis (ALS), a form of motor neurone disease, which can cause muscle degeneration leading to muscle weakness.
For more information, see Section 1. Why am I using Teglutik®? in the full CMI.
2. What should I know before I use Teglutik®?
Do not use if you have ever had an allergic reaction to Teglutik® or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use Teglutik®? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with Teglutik® and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use Teglutik®?
- The recommended daily dose is 20mL (10mL every 12 hours).
More instructions can be found in Section 4. How do I use Teglutik®? in the full CMI.
5. What should I know while using Teglutik®?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Drinking alcohol |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using Teglutik®? in the full CMI.
6. Are there any side effects?
The most common side effects are stomachache, nausea or vomiting, headache, joint stiffness, skin problems eg. rash, flaking skin, dizziness, sleepiness, and weakness or loss of strength. Serious side effects which may require urgent medical attention may include irregular or fast heartbeat, frequent infections such as fever, severe chills, sore throat or mouth ulcers, swelling of the hands, feet or legs, tingling sensations around the mouth, shortness of breath or difficulty breathing, severe upper stomach pain (often with nausea and vomiting), itchy skin or yellow skin, or if you start to bleed or bruise easily.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
Teglutik® Oral Suspension (Tee-gloo-tick)
Active ingredient(s): riluzole (Ril-you-zol)
Consumer Medicine Information (CMI)
This leaflet provides important information about using Teglutik®. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using Teglutik®.
Where to find information in this leaflet:
1. Why am I using Teglutik®?
2. What should I know before I use Teglutik®?
3. What if I am taking other medicines?
4. How do I use Teglutik®?
5. What should I know while using Teglutik®?
6. Are there any side effects?
7. Product details
1. Why am I using Teglutik®?
Teglutik® contains the active ingredient riluzole. Teglutik® is Schedule 4 (S4): Prescription Only Medicine.
Teglutik® is used to treat people with amyotrophic lateral sclerosis (ALS), a form of motor neurone disease, which can cause muscle degeneration leading to muscle weakness.
2. What should I know before I use Teglutik®?
Warnings
Do not use Teglutik® if:
- you are allergic to riluzole or any of the ingredients listed at the end of this leaflet.
- you have liver disease
- you are pregnant or intend to become pregnant
- you are breastfeeding or intend to breastfeed
- Always check the ingredients to make sure you can use this medicine.
Children and adolescents
This medicine should not be given to children and adolescents under 18 years of age because there is no experience with the use of this medicine in children.
Check with your doctor if you:
- have any other medical conditions such as:
- liver disease;
- kidney disease;
- lung disease - take any medicines for any other condition
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
This medicine is not recommended to be used during pregnancy.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
It is not known whether it passes into breast milk. Your doctor will discuss the risks and benefits of taking Teglutik® if you are breastfeeding or planning to breastfeed.
Intolerance to sugars
If you have been told by your doctor that you have an intolerance to some sugars, contact your doctor before taking this medicinal product.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interfere with Teglutik® and affect how it works. These include:
- theophylline - a medicine used to treat asthma
- amitriptyline - a medicine used to treat depression
- tacrine - a medicine used in patients with Alzheimer's Disease
- some types of antibiotics eg. rifampicin and quinolones
- omeprazole - a medicine used to treat gastric ulcers.
- some medicines used to treat depression eg. Clomipramine and fluvoxamine
- diazepam - a medicine for sedation.
- diclofenac - a medicine used to reduce pain and inflammation.
These medicines may be affected by Teglutik® or may affect how well it works. You may need to use different amounts of your medicine, or you may need to take different medicines. Your doctor or pharmacist will advise you.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect Teglutik®.
4. How do I use Teglutik®?
How much to take
- The recommended daily dose is 20mL (10mL every 12 hours).
- Follow the instructions provided and use Teglutik® until your doctor tells you to stop.
When to take Teglutik®
- Teglutik®should be taken by mouth every 12 hours, at the same time of the day each day (for example, in the morning and evening).
- Teglutik® should not be taken immediately before or after meals, especially meals which may contain food high in fat.
- Teglutik® may not work as well if it is taken at the same time as your meals.
How to take Teglutik®
- Follow all directions given to you by your doctor or pharmacist carefully.
- If you do not understand the instructions in this leaflet, ask your doctor or pharmacist for help.
- Teglutik® should be taken by mouth or via a Percutaneous Endoscopic Gastrostomy (PEG)
- Teglutik® is administered by means of graduated dosing syringe.
Method of administration:
- The bottle of Teglutik® should be gently shaken for at least 30 seconds by rotating the bottle by 180°. You will know when the liquid turns slightly brown.
- Open the bottle: press the cap and turn it anticlockwise (figure 1).
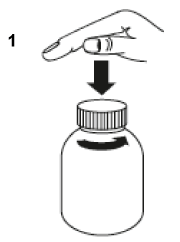
- Take the syringe, remove the tip and insert the syringe in the adaptor opening (figure 2). Turn the bottle upside down (figure 3).
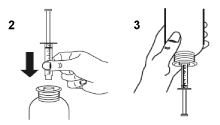
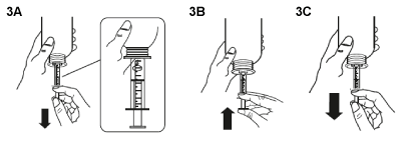
- Fill the syringe with a small amount of Teglutik® by pulling the plunger down (figure 3A), then push the piston upward in order to remove any possible bubble (figure 3B). Pull the piston down to the graduation mark corresponding to the quantity in milliliters (mL) prescribed by your doctor (figure 3C).
- Turn the bottle the right way up (figure 4A). Remove the syringe from the adaptor (figure 4B).
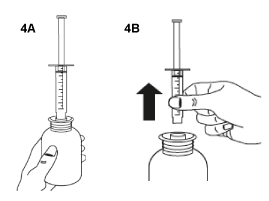
- Administer orally the whole content of the syringe.
Dilution in water is not necessary. - Close the bottle with the plastic screw cap.
- Wash the syringe with water only and re-assemble it with its tip cap once dried (figure 5).
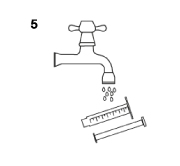
- For PEG administration, the tubing should be washed with 10 mL of water before and after administration of Teglutik®.
If you forget to use Teglutik®
Teglutik® should be used regularly at the same time each day. If you miss or forget your dose at the usual time, take it as soon as you remember, and then go back to taking your medicine as you would normally.
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Do not take a double dose to make up for the dose you missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you use too much Teglutik®
If you think that you have used too much Teglutik®, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using Teglutik®?
Things you should do
- Tell any doctor, dentist or pharmacist you visit that you are using Teglutik®
- Tell your doctor or pharmacist if you are about to be started on any new medicine that you are taking Teglutik®.
- Tell your doctor or dentist that you are taking Teglutik® if you plan to have surgery that needs a general anesthetic.
- Tell your doctor if you smoke and how much coffee you drink. Nicotine and caffeine may affect the amount of Teglutik® in your body.
- During your treatment with Teglutik®your doctor will do some blood tests from time to time to check for any possible signs of liver damage.
Call your doctor straight away if you:
- become pregnant while you are taking Teglutik®
Things you should not do
- Do not stop taking this medicine, or lower the dosage, without checking with your doctor.
- Do not take more than the recommended dose unless your doctor tells you to.
- Do not give your medicine to anyone else, even if they have the same condition as you.
- Do not use this medicine to treat any other complaints unless your doctor tells you to.
- Do not use this medicine if the bottle shows signs of tampering.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how Teglutik® affects you.
Teglutik® may cause drowsiness or dizziness in some people. Make sure you know how you react to it before you drive a car, operate machinery, or do anything else that could be dangerous if you are dizzy.
Drinking alcohol
Tell your doctor if you drink alcohol.
Looking after your medicine
- Store Teglutik® inside the box, until it is time to take it.
- If you take Teglutik® out of the box or bottle it may not keep well.
- Store in a cool, dry place where the temperature stays below 25°C.
- Do not store Teglutik® in the bathroom or near a sink.
- Do not leave it in the car or on window sills.
- Heat and dampness can destroy some medicines.
Follow the instructions in the carton on how to take care of your medicine properly.
Keep it where children cannot reach it.
- A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
When to discard your medicine
- If your doctor tells you to stop taking Teglutik®
- If the expiry date on the carton or Teglutik® bottle has passed.
- If 15 days has passed after first opening Teglutik®.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
Stomach related:
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
General complaints:
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Liver complaints:
| If any of the following happen, stop taking this medicine and tell your doctor immediately, or go to Accident and Emergency at your nearest hospital. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What Teglutik® contains
| Active ingredient (main ingredient) | The active substance is riluzole. 1 mL of Teglutik® contains 5 mg of riluzole. |
| Other ingredients (inactive ingredients) |
|
| Potential allergens |
|
Do not take this medicine if you are allergic to any of these ingredients.
What Teglutik® looks like
Teglutik® is presented as a slightly brown, opaque homogeneous oral suspension after being manually gently shaken.
Teglutik® is available in a carton containing a bottle of 250 mL or 300 mL, or in a multipack consisting of 2 single cartons each with one bottle of 250 mL. Each carton is supplied with a plastic graduated oral dosing syringe. The syringe barrel is graduated in millilitres up to 10 mL. Not all pack sizes may be marketed.
Who distributes Teglutik®
Asteri Pharma Pty Ltd
6 Lux Way
Brunswick, Victoria 3056
Australia
Telephone: 1800 175 659
Website: www.asteripharma.com
This leaflet was prepared in January 2026.
Brand Information
| Brand name | Teglutik Oral Suspension |
| Active ingredient | Riluzole |
| Schedule | S4 |
MIMS Revision Date: 01 March 2026
1 Name of Medicine
Riluzole.
2 Qualitative and Quantitative Composition
Each 1 mL of oral suspension contains 5 mg of riluzole.
Riluzole is a white to slightly yellow powder. It is very slightly soluble in water and 0.1 N sodium hydroxide, sparingly soluble in 0.1 N hydrochloric acid; and very soluble in methanol, acetone, acetonitrile, dichloromethane and dimethyl sulfoxide.
1 mL of oral suspension contains 400 mg of sorbitol (equivalent to 571.43 mg of sorbitol solution (70% w/w)). For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
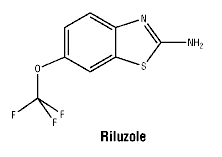
Teglutik is an oral suspension containing riluzole, a benzothiazole. Chemical name: 6-(trifluoromethoxy)-2-benzothiazolamine It is a slightly brown, opaque homogenous suspension after manually shaken.
4 Clinical Particulars
4.1 Therapeutic Indications
Riluzole is indicated for the treatment of patients with amyotrophic lateral sclerosis (ALS).
4.2 Dose and Method of Administration
Dosage. The recommended dose is 10 mL (equivalent to 50 mg riluzole) two times a day. The maximum recommended daily dose is 100 mg (50 mg every 12 hours). No significant increase in benefit can be expected from higher daily doses.
Due to the reduction in absorption observed when administered with high fat meals, riluzole should not be taken with a fat containing meal.
Method of administration. The suspension can be given per oral administration. Dilution with liquids is not necessary.
The suspension is administered by means of graduated dosing syringe orally or via a Percutaneous Endoscopic Gastrostomy (PEG).
The suspension must be manually gently shaken for at least 30 seconds by rotating the bottle by 180° and the homogeneity should be visually verified.
Open the bottle, connect the dosing syringe to the bottle syringe-adapter, invert the bottle and, by maintaining the bottle in the inverted position, slowly withdraw the suspension volume corresponding to the recommended dose (i.e. 10 mL corresponds to 50 mg of riluzole).
After the administration of the suspension, thoroughly wash the syringe with tap water. The PEG tube should be washed with tap water before and after administration of the suspension.
4.3 Contraindications
Patients who have a history of severe hypersensitivity reactions to riluzole or any of the excipients.
Patients who have a hepatic disease or hepatic impairment (baseline transaminases greater than 3 times the upper limit of normal).
Patients who are pregnant or lactating.
This product contains sorbitol.
Patients with rare hereditary problems of fructose intolerance should not take this medicine (see Section 4.4 Special Warnings and Precautions for Use).
4.4 Special Warnings and Precautions for Use
Use in hepatic impairment. Riluzole is contraindicated in patients with hepatic disease or hepatic impairment (baseline transaminases greater than 3 times the upper limit of normal).
Riluzole should be prescribed with care in patients with a history of abnormal liver function, or in patients with slightly elevated serum transaminase (ALT/SGPT; AST/SGOT up to 3 times the ULN), bilirubin and/or gamma-glutamyl transferase (GGT) levels. Baseline elevations of several liver function tests (especially elevated bilirubin) should preclude the use of riluzole.
Elevations of alanine-aminotransferase (ALT) levels to more than 3 times the upper limit of the normal range (ULN) were observed in about 10% of the patients treated with riluzole compared to 3.7% in the placebo group; levels increased to more than 5 times the ULN in about 3% of the patients treated with riluzole compared to 2% of the placebo treated patients. The increases in ALT usually appeared within 3 months after the start of therapy with riluzole; they were usually transient and levels returned to below 2 times the ULN after 2 to 6 months while treatment was continued. These increases were rarely associated with jaundice. In patients with increases in ALT to more than 5 times the ULN, treatment was discontinued and the levels returned to less than 2 times the ULN within 2 to 4 months.
Because of risks of hepatitis, serum transaminases, including ALT, should be measured before and during therapy with riluzole. ALT should be measured every month during the first 3 months of treatment, every 3 months during the remainder of the first year, and periodically thereafter. ALT levels should be measured more frequently in patients who develop elevated ALT levels.
Riluzole should be discontinued if the ALT levels increase to five times the ULN. There is no experience with dose reduction or rechallenge in patients who have developed an increase of ALT to 5 times ULN. Readministration of riluzole to patients in this situation cannot be recommended.
Use in renal impairment. Riluzole should be used with caution in patients with renal insufficiency.
Hereditary fructose intolerance. The product contains liquid sorbitol (E420) therefore patients with rare hereditary problems of fructose intolerance should not take this medicine.
Neutropenia. There have been three reports (3/5000) of marked neutropenia where absolute neutrophil count was less than 500/mm3. See Section 4.8 Adverse Effects (Undesirable Effects). Patients should be warned to report any febrile illness to their physicians. The report of a febrile illness should prompt physicians to check white blood cell counts and to discontinue riluzole in case of neutropenia.
Interstitial lung disease. Cases of interstitial lung disease have been reported in patients treated with riluzole, some of them were severe (see Section 4.8 Adverse Effects (Undesirable Effects)). If respiratory symptoms develop such as dry cough and/or dyspnea, chest radiography should be performed, and in case of findings suggestive of interstitial lung disease (e.g. bilateral diffuse lung opacities), riluzole should be discontinued immediately. In the majority of reported cases, symptoms resolved after drug discontinuation and symptomatic treatment.
Use in elderly. See Section 5.2 Pharmacokinetic Properties.
Paediatric use. The safety and effectiveness of riluzole in any neurodegenerative process occurring in children or adolescents have not been established.
4.5 Interactions with Other Medicines and Other Forms of Interactions
There have been no clinical studies to evaluate the drug interactions of riluzole with other drugs. Experiments on mice and rats indicated that riluzole potentiated the hypnotic effects of hexobarbitone and chlorpromazine.
The metabolism of riluzole is mostly hepatic and consists of cytochrome P450-dependent hydroxylation and glucuronidation. There is marked inter-individual variability in the clearance of riluzole, probably attributable to variability of CYP 1A2 activity, the principal isozyme involved in N-hydroxylation. In vitro studies using liver microsomes show that hydroxylation of the primary amine group producing N-hydroxyriluzole is the main metabolic pathway in humans. In humans, cytochrome P450 1A2 is the principal isozyme involved in N-hydroxylation. In vitro studies predict that CYP 2D6, CYP 2C19, CYP 3A4, and CYP2E1 are unlikely to contribute significantly to riluzole metabolism in humans.
Effect of riluzole on the metabolism of other drugs. Potential interactions may occur when riluzole is given concurrently with other agents which are also metabolized primarily by CYP 1A2 (e.g. theophylline, caffeine and tacrine). It is not known whether riluzole has any potential for enzyme induction in humans.
Effect of other drugs on riluzole metabolism. Potential interactions may occur when riluzole is given concurrently with other agents that affect CYP 1A2 activity. Potential inhibitors of CYP 1A2 (e.g. caffeine, diclofenac, diazepam, nicergoline, clomipramine, imipramine, fluvoxamine, phenacetin, theophylline, amitriptyline and quinolones) could decrease the rate of riluzole elimination, while inducers of CYP 1A2 (e.g. cigarette smoke, charcoal-broiled food, rifampicin and omeprazole) could increase the rate of riluzole elimination.
4.6 Fertility, Pregnancy and Lactation
Effect on fertility. Riluzole impaired fertility when administered to male and female rats prior to mating and during mating at an oral dose of 15 mg/kg (approximately 13 times human exposure at the maximum recommended clinical dose of 100 mg, based on AUC).
Use in pregnancy. (Category B3)
In the pregnant rat, the transfer of 14C-riluzole across the placenta to the foetus has been detected. There was no evidence of embryotoxicity or teratogenicity in the offspring of rats or rabbits following maternal treatment with riluzole during organogenesis at oral doses of up to 27 and 60 mg/kg/day respectively, corresponding to plasma exposures (based on AUC) 61 and 18 times higher than those anticipated in clinical use. However, foetal growth and development were slightly retarded, possibly as a consequence of maternal toxicity. Foetal growth was not affected following maternal exposure to riluzole at levels approximately 6 to 8-fold higher (based on AUC) than those anticipated in clinical use.
When administered to rats prior to and during mating (males and females) and throughout gestation and lactation (females), riluzole produced adverse effects on pregnancy (decreased implantations) and offspring viability and growth at an oral dose of 15 mg/kg (approximately 13 times human exposure at the maximum recommended clinical dose of 100 mg, based on AUC).
There are no adequate and well-controlled studies in pregnant women. Riluzole must not be used in pregnant women.
Use in lactation. 14C-riluzole and/or its metabolites were detected in the milk of lactating rats at levels 2.5-fold higher than those appearing in maternal plasma. There was an increased incidence of postnatal mortality in the offspring of rats treated with riluzole during the peri-natal period at oral doses of 15 mg/kg/day, which represents exposure (on the basis of AUC) to levels 13-fold higher than those anticipated in clinical use. It is not known whether riluzole is excreted in human milk; therefore, women should not breast-feed during treatment with riluzole.
4.7 Effects on Ability to Drive and Use Machines
Patients should be warned about the potential for dizziness, vertigo or somnolence, and advised not to drive or operate machinery if these symptoms occur.
4.8 Adverse Effects (Undesirable Effects)
Clinical trials. In Phase III studies conducted in North America and Europe, the most frequent side effects related to riluzole were asthenia, nausea, and elevations in liver function tests. Table 1 includes all the adverse events that occurred at a frequency of 1% or more among ALS patients receiving riluzole 100 mg/day:
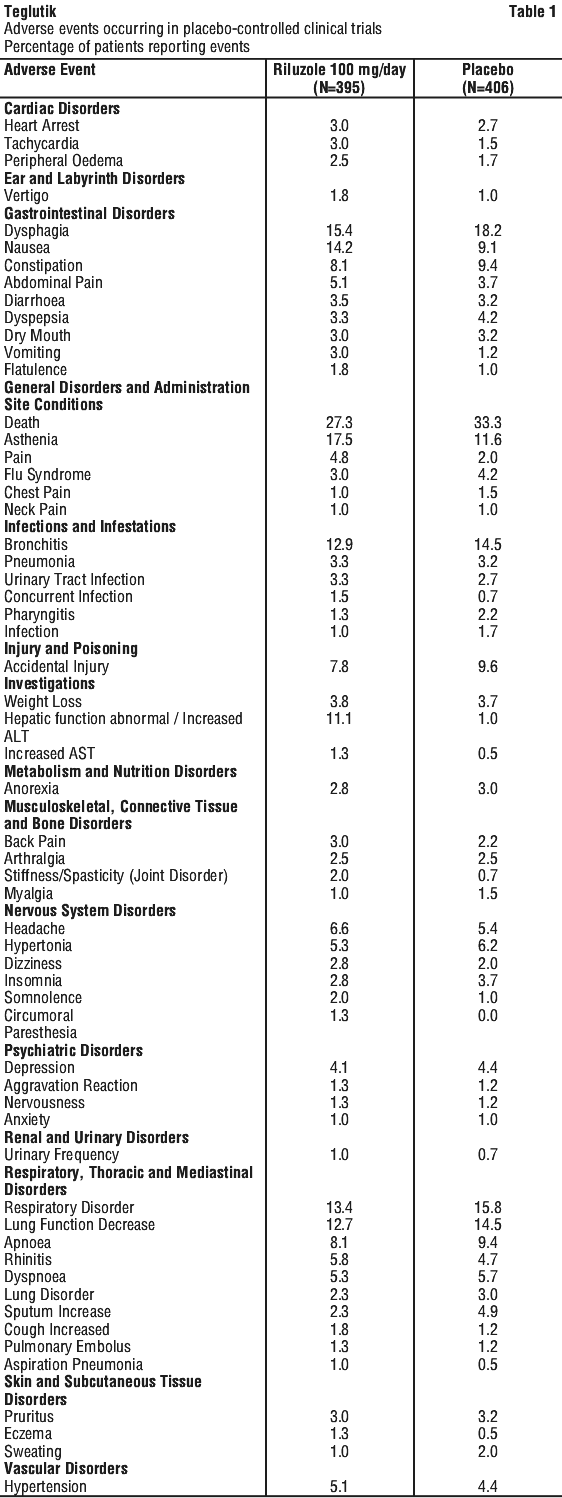
The following is a list of adverse reactions reported from clinical trials and post marketing studies with an incidence of less than 1%:
Uncommon 0.1 - 1%; rare 0.01 - 0.1%; very rare < 0.01%; not known (cannot be estimated from the available data).
Cardiac disorders. Rare: angina unstable, atrial fibrillation, cardiac failure.
Very Rare: arrhythmia.
Gastrointestinal disorders. Uncommon: pancreatitis.
Rare: gastrointestinal disorder, gastric ulcer, gastrointestinal haemorrhage, gastrointestinal irritation, melaena.
General disorders and administration site conditions. Rare: condition aggravated, malaise, weakness, pyrexia.
Very rare: anaphylactoid reaction.
Hepato-biliary disorders. Rare: hepatitis, jaundice, hepatocellular damage.
Immune system disorders. Uncommon: anaphylactoid reaction, angioedema.
Rare: hypersensitivity.
Laboratory investigations. Rare: gamma-glutamyltransferase increased, liver function tests abnormal, transaminase increased, blood bilirubin increased, blood alkaline phosphatase increased, haematocrit decreased, blood creatine phosphokinase increased, glycosuria present, haemoglobin decreased, leukocyte count decreased, platelet count decreased.
Metabolism and nutrition disorders. Rare: dehydration.
Very rare: hyponatraemia.
Nervous system disorders. Very rare: amnesia.
Psychiatric disorders. Rare: motor dysfunction, paraesthesia nec, completed suicide, confusion, delirium, hallucination, personality change due to a general medical condition.
Respiratory, thoracic and mediastinal disorders. Uncommon: respiratory failure (exc neonatal), interstitial lung disease (see Section 4.4 Special Warnings and Precautions for Use).
Rare: asphyxia, respiratory distress.
Skin and subcutaneous tissue disorders. Rare: dermatitis.
Very rare: angioedema.
Blood and lymphatic system disorders. Uncommon: anaemia.
Rare: erythropenia, leucopoenia, thrombocytopenia.
Very rare: neutropenia - among approximately 5000 patients given riluzole for ALS, there were three cases of marked neutropenia (absolute neutrophil count less than 500/mm3), all seen within the first 2 months of riluzole treatment. In one case, neutrophil counts rose on continued treatment. In a second case, counts rose after therapy was stopped. A third case was associated with marked anaemia and the aetiology is uncertain.
4.9 Overdose
Neurological and psychiatric symptoms, acute toxic encephalopathy with stupor, coma and methemoglobinaemia have been observed in isolated cases. Severe methemoglobinaemia may be rapidly reversible after treatment with methylene blue. In case of overdose, treatment is symptomatic and supportive.
For information on the management of overdose, contact the Poisons Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action. The aetiology and pathogenesis of amyotrophic lateral sclerosis (ALS) are not known, although a number of hypotheses have been advanced. One hypothesis is that motor neurones made vulnerable through either genetic predisposition or environmental factors, are injured by glutamate. In some cases of familial ALS, enzyme superoxide dismutase has been found to be defective.
The mechanism of action of riluzole has not been completely elucidated but evidence to date suggests that it may involve inactivation of voltage dependent sodium channels and impairment of glutamatergic neurotransmission.
There are no validated animal models of ALS in which to test riluzole. Riluzole has been shown to cross the blood brain barrier and to possess neuroprotective properties in various in vivo experimental models of neuronal injury known to involve excitotoxic mechanisms, such as cerebral ischemia. In vitro, riluzole protects cultured rat motorneurones from the excitotoxic effects of glutamic acid and prevents the death of cortical neurones induced by anoxia. In healthy volunteers at therapeutic doses, riluzole has been shown to protect to some extent against the hypobaric hypoxia induced at an equivalent altitude of 5000 m. Also, riluzole moderately reduces the cerebral metabolic rate of glucose as shown by PET-scan.
Due to its blockade of glutamatergic neurotransmission, riluzole also has myorelaxant and sedative properties in animal studies at doses of 30 mg/kg (about 20 times the human recommended daily dose) and anticonvulsant properties at doses of 2.5 mg/kg (about 2 times the human recommended daily dose).
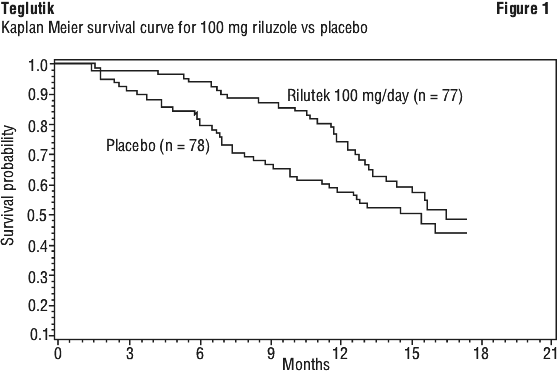
Clinical trials. Two multinational, multicenter, double-blind, parallel group trials have demonstrated that Teglutik extends survival for patients with ALS regardless of the onset type. It is also concluded that the survival benefit is maintained.
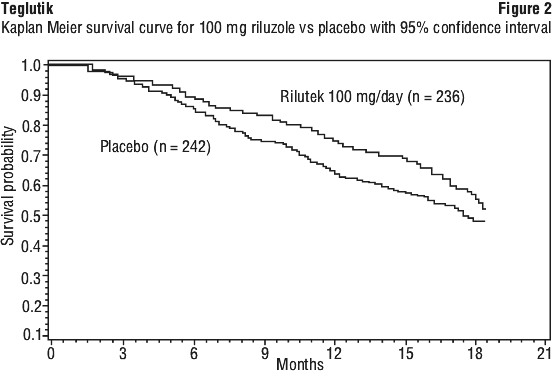
In a first trial, 155 patients were randomised to riluzole 100 mg/day (50 mg twice daily) or placebo and were followed-up for 12 to 21 months. While there was no change from baseline in the functional evaluation, survival was significantly prolonged for patients who received riluzole as compared to patients who received placebo. The median survival time was 17.7 months versus 14.9 months for riluzole and placebo respectively. See Figure 1.
Riviere et al. (1998) analysed extended survival in ALS patients treated with riluzole in this study. Post hoc analysis suggested that the patients receiving riluzole remained in the milder health states longer (p < 0.05, Cox model). Patients with advanced disease were less responsive.
In a double-blind placebo-controlled trial designed to assess the efficacy and safety of riluzole in Japanese patients, patients were randomised to riluzole 100 mg/day (50 mg twice daily) or placebo and were followed-up for 18 months. In this study, the efficacy was assessed on inability to walk alone, loss of upper limb function, tracheostomy, need for artificial ventilation, gastric tube feeding or death. Tracheostomy-free survival in patients treated with riluzole did not differ significantly from placebo. Due to the low incidence of ALS in Japan, and for practical reasons, the study was limited to 100 patients per treatment group. The small size of this study resulted in a lack of statistical power to detect a significant difference between riluzole and placebo.
Meta analysis, including this study and those described above, showed a less striking effect of survival for riluzole as compared to placebo although the differences remained statistically significant.
A Cochrane Review of data from the two pivotal studies (first trial and dose ranging trial) found that there was a significant difference in percent mortality at 12 months between riluzole 100 mg/day and placebo groups. Results were expressed as odds ratios (OR) and 95% CI for continuous variables. With regards to the primary outcome (mortality at 12 months) the OR for the combined studies was 0.57 (95% CI 0.41 to 0.80, p=0.001). There was no evidence of heterogeneity (Chi-square, p=0.58). Overall there was a 23% reduction in risk of death in those patients receiving riluzole (p=0.0509).
A United Kingdom National Institute for Clinical Excellence (NICE) Review of the clinical effectiveness of riluzole found that it was effective in the treatment of ALS. In a meta-analysis which included data from the two pivotal studies and the compassionate study, it was found that for tracheostomy-free survival over 18 months the hazard ratio was 0.83 (95% CI 0.69-0.99). The report concluded that there was evidence of a modest benefit for patients taking riluzole.
5.2 Pharmacokinetic Properties
The pharmacokinetics of riluzole have been evaluated in healthy male volunteers after single oral administration of 25 to 300 mg and after multiple-dose oral administration of 25 to 100 mg bid. Plasma levels increase linearly with the dose and the pharmacokinetic profile is dose-independent. Steady-state plasma levels are reached within 3 to 8 days.
Absorption. Riluzole is rapidly absorbed after oral administration with maximal plasma concentrations occurring within 60 to 90 minutes (Cmax = 173 ± 72 (sd) nanogram/mL). About 90% of the dose is absorbed and the absolute bioavailability is 60 ± 18%. With multiple dose administration (10 day treatment at 50 mg riluzole bid), unchanged riluzole accumulates in plasma by about 2 fold and steady-state is reached in less than 5 days.
The rate and extent of absorption is reduced when riluzole is administered with high-fat meals (decrease in Cmax of 44%, decrease in AUC of 17%).
In a comparative bioavailability study the AUC0-α of riluzole 50 mg tablets and of Teglutik (riluzole) 50 mg/10 mL oral suspension were equivalent. (Ratio: 106.46%; 90% CI: 96.68 - 117.23%). Riluzole is more rapidly absorbed after the administration of oral suspension (Tmax approximately 30 minutes) with a mean Cmax approximately 20% higher than after the administration of riluzole tablets (Ratio: 122.32%; 90% CI: 103.28-144.88%) (see Section 4.8 Adverse Effects (Undesirable Effects)).
Distribution. Riluzole is extensively distributed throughout the body and has been shown to cross the blood brain barrier. The volume of distribution of riluzole is about 245 ± 69 L (3.4 L/kg). Riluzole is about 97% protein bound and it binds mainly to serum albumin and to lipoproteins.
Metabolism. Riluzole is extensively metabolized to six major and a number of minor metabolites, not all of which have been identified. Some metabolites appear pharmacologically active in in vitro assays. The metabolism of riluzole is mostly hepatic and consists of cytochrome P450-dependent hydroxylation and glucuronidation.
There is marked interindividual variability in the clearance of riluzole, probably attributable to variability of CYP 1A2 activity, the principal isozyme involved in N-hydroxylation.
In vitro studies using liver microsomes show that hydroxylation of the primary amine group producing N-hydroxyriluzole is the main metabolic pathway in human, monkey, dog and rabbit. In humans, cytochrome P450 1A2 is the principal isozyme involved in N-hydroxylation. In vitro studies predict that CYP 2D6, CYP 2C19, CYP 3A4 and CYP 2E1 are unlikely to contribute significantly to riluzole metabolism in humans. Whereas direct glucuroconjugation of riluzole (involving the glucurotransferase isoform UGT-HP4) is very slow in human liver microsomes, N-hydroxyriluzole is readily conjugated at the hydroxylamine group resulting in the formation of O-(> 90%) and N-glucuronides.
Elimination. The elimination half-life ranges from 9 to 15 hours. Riluzole is eliminated mainly in the urine. The overall urinary excretion accounts for about 90% of the dose. Glucuronides accounted for more than 85% of the metabolites in the urine. Only 2% of a riluzole dose was recovered unchanged in the urine.
Special populations. Elderly. The pharmacokinetics of riluzole in elderly subjects were compared to young healthy subjects and no clinically significant differences were found.
Gender. No gender effect on the pharmacokinetics of riluzole was found, however CYP 1A2 activity has been reported to be lower in women than in men and thus a higher blood concentration of riluzole and its metabolites is possible in women.
A comparative bioavailability study has been conducted between Teglutik oral suspension and riluzole tablets. The results showed, as reported also in the literature, a higher exposure in female subjects in terms of Cmax (~25%) and AUC (~53%) of Teglutik oral suspension compared to male subjects. However, no relevant clinical impact is expected.
Smoking. Cigarette smoking is known to induce CYP 1A2 and thus it is possible that patients who smoke may eliminate riluzole faster. There is no information available on the effect or need for dosage adjustment.
Race. Clearance of riluzole in native Japanese subjects was found to be 50% lower compared to Caucasian subjects (after normalizing for body weight). Although it is not clear if this difference is due to genetic or environmental factors (e.g. smoking, alcohol, coffee and dietary preferences) it is possible that Japanese subjects may possess a lower capacity (oxidative and/or conjugative) for metabolising riluzole. There are no studies, however, of lower doses in Japanese subjects.
Paediatric. The safety and efficacy of riluzole in children has not been studied.
Renal impairment. Study results showed that the pharmacokinetic profile of a single dose of riluzole is similar between patients with moderate or severe chronic renal insufficiency (creatinine clearance between 10 and 50 mL/min) and healthy subjects. A multiple dose study in renally impaired patients has not been performed.
Hepatic impairment. The AUC of riluzole after a single oral dose of 50 mg increases by about 1.7 fold in patients with mild chronic liver insufficiency and by about 3 fold in patients with moderate chronic liver insufficiency. See Section 4.3 Contraindications; Section 4.4 Special Warnings and Precautions for Use.
5.3 Preclinical Safety Data
Carcinogenicity. Two long term (2 years) carcinogenicity studies have been completed in rats and mice. Riluzole showed no evidence of carcinogenic potential in rats and mice treated orally for 2 years at doses of 10 and 20 mg/kg/day, respectively. These doses were approximately 0.85 times the recommended maximum dose of 100 mg daily, on a mg/m2 basis.
There was no evidence of a genotoxic potential in standard assays for gene mutations (microbial mutagenicity test, mouse lymphoma assay in L5178Y cells) and chromosomal damage (chromosomal aberrations in human lymphocytes in vitro, rat cytogenetic assay in vivo and mouse micronucleus assay).
6 Pharmaceutical Particulars
6.1 List of Excipients
The oral suspension also contains sorbitol solution (70% w/w) (non-crystallising), aluminium magnesium silicate, xanthan gum, saccharin sodium, antifoam AF emulsion Q7-2587, sodium lauryl sulfate, ceteareth-25, and purified water.
6.2 Incompatibilities
See Section 4.5 Interactions with Other Medicines and Other Forms of Interactions.
6.3 Shelf Life
Teglutik oral suspension has a shelf life of 3 years when stored below 25°C.
6.4 Special Precautions for Storage
After the first opening: 15 days, without any special storage conditions.
6.5 Nature and Contents of Container
1 mL of oral suspension contains 5 mg of riluzole.
The suspension is supplied in an amber glass bottle equipped with a LDPE syringe-adapter and closed by means of a white-white HDPE child-proof screw cap.
Pack sizes of one or two bottles of 250 mL of Teglutik 5 mg/mL oral suspension.
Pack size of one bottle of 300 mL of Teglutik 5 mg/mL oral suspension.
The bottle is packed with a plastic graduated oral dosing syringe. The syringe barrel is graduated in millilitres up to 10 mL.
Not all pack sizes may be marketed.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
7 Medicine Schedule (Poisons Standard)
Schedule 4 (S4): Prescription Only Medicine.
Date of First Approval
21 August 2018
Date of Revision
06 January 2026
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.