Truqap
Brand Information
| Brand name | Truqap |
| Active ingredient | Capivasertib |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Truqap.
Summary CMI
TRUQAP®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
▼ This medicine is new or being used differently. Please report side effects. See the full CMI for further details.
1. Why am I using TRUQAP?
TRUQAP contains the active ingredient capivasertib. TRUQAP is used in combination with another medicine called fulvestrant to treat adults who have hormone receptor (HR) positive, human epidermal growth factor receptor 2 (HER2) negative breast cancer that is advanced or that has spread to other parts of the body and whose cancer is not responding to other anti-hormonal based therapies.
For more information, see Section 1. Why am I using TRUQAP? in the full CMI.
2. What should I know before I use TRUQAP?
Do not use if you have ever had an allergic reaction to capivasertib or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use TRUQAP? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with TRUQAP and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use TRUQAP?
- The usual dose of TRUQAP is two 200 mg tablets taken twice daily for 4 days followed by 3 days off treatment. However your doctor may prescribe a different dose if you are taking certain medicines or if you experience certain side effects.
- Continue using TRUQAP for as long as your doctor tells you to.
More instructions can be found in Section 4. How do I use TRUQAP? in the full CMI.
5. What should I know while using TRUQAP?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using TRUQAP? in the full CMI.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. These include nausea, vomiting, tiredness, mouth sores or ulcers. However, some serious side effects may need medical attention. These can include diarrhoea, high blood sugar levels, severe blistering, peeling or scaling of skin or skin reactions.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
▼ This medicine is subject to additional monitoring. This will allow quick identification of new safety information. You can help by reporting any side effects you may get. You can report side effects to your doctor, or directly at www.tga.gov.au/reporting-problems.
TRUQAP®
Active ingredient: capivasertib
Consumer Medicine Information (CMI)
This leaflet provides important information about using TRUQAP. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using TRUQAP.
Where to find information in this leaflet:
1. Why am I using TRUQAP?
2. What should I know before I use TRUQAP?
3. What if I am taking other medicines?
4. How do I use TRUQAP?
5. What should I know while using TRUQAP?
6. Are there any side effects?
7. Product details
1. Why am I using TRUQAP?
TRUQAP contains the active ingredient capivasertib.
Capivasertib belongs to a group of medicines called AKT inhibitors. TRUQAP works by blocking the effects of proteins called AKT Kinases. These proteins help cancer cells to grow and multiply. By blocking their action, TRUQAP can reduce growth and spread of the cancer and help to destroy cancer cells.
TRUQAP is used in combination with another medicine called fulvestrant to treat adults who have hormone receptor (HR) positive, human epidermal growth factor receptor 2 (HER2) negative breast cancer that is advanced or that has spread to other parts of the body and whose cancer is not responding to other anti-hormonal based therapies.
2. What should I know before I use TRUQAP?
Warnings
Do not use TRUQAP if:
- You are allergic to capivasertib, or any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin or
- you may feel faint
Check with your doctor if you:
- have or have ever had high levels of sugar in your blood or diabetes (or signs of increased sugar levels, such as excessive thirst and dry mouth, needing to pass urine more often than usual, producing greater amounts of urine than usual, increased appetite with weight loss).
- have any current infections.
- have diarrhoea or loose stool.
- have rash or other skin disorders.
- have kidney problems or high levels of creatinine or uric acid in your blood.
- have liver problems.
- have any other medical conditions
- take any medicines for any other condition
- have allergies to any other medicines, foods, preservatives or dyes.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Tell your doctor if you are pregnant or if you (male or female) are planning to have a baby.
If you are pregnant or plan to become pregnant, you should not take TRUQAP. Before taking TRUQAP, tell your doctor if you are pregnant, think you may be pregnant or planning to have a baby as TRUQAP may harm your unborn baby.
If you are a woman who could become pregnant, your doctor will ask you to provide a negative pregnancy test prior to starting treatment and may advise you to perform a pregnancy test during your treatment. If you are able to become pregnant, you should use effective birth control during treatment with TRUQAP and for 4 weeks after the last dose.
Male patients should use condoms and effective birth control during treatment and for 16 weeks after the last dose.
If you or your partner do become pregnant during the treatment tell your doctor right away.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
For the safety of your baby, you should not breast-feed during the treatment with TRUQAP.
Children and Adolescents
The safety and efficacy of TRUQAP has not been established in children or adolescents. Do not give this medicine to children or adolescents aged less than 18 years.
Monitoring before and during treatment with TRUQAP
It is important that you have your blood sugar monitored during treatment. Your doctor will perform blood tests to measure your blood sugar levels before you start taking TRUQAP and during treatment. Based on the results, your doctor may decide to temporarily interrupt treatment with TRUQAP or reduce the dose of TRUQAP or decide to stop the treatment.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking or have recently been taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some products may interfere with TRUQAP and affect how it works.
Taking TRUQAP with certain other medicines may affect how TRUQAP works and can cause side effects. Some medicines can affect the levels of TRUQAP in your body. Also, TRUQAP can affect the way some other medicines work. Some medicines may increase the risk of side effects of TRUQAP.
These include:
- antibiotics for treating bacterial infections such as clarithromycin, rifampicin
- medicines used to treat fungal infections such as ketoconazole, itraconazole and voriconazole
- medicines for epilepsy such as carbamazepine and phenytoin
- St John's Wort, a herbal medicine used to treat depression
- anti-viral medicines such as saquinavir, ritonavir and lopinavir
- bosentan, a medicine used to treat high blood pressure in the lungs
- simvastatin a medicine used to treat high levels of cholesterol
- medicines used to suppress the immune system such as ciclosporin and tacrolimus
- medicines used to treat cancer such as ceritinib, tucatinib.
Avoid drinking large amounts of grapefruit juice while taking TRUQAP as it may increase side effects of TRUQAP.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are or have been taking and if these affect TRUQAP.
4. How do I use TRUQAP?
How much to take
The usual dose of TRUQAP is 400 mg (two 200 mg tablets) taken orally twice daily approximately 12 hours apart (total daily dose of 800 mg) for 4 days followed by 3 days off treatment. Your doctor may prescribe a different dose if you are taking certain medicines that may interact with TRUQAP or if you experience certain side effects while you are taking TRUQAP.
Swallow TRUQAP tablets whole with water. Do not chew, crush, dissolve or divide the tablets. TRUQAP tablets may be taken with or without food.
If you vomit, do not take an additional dose. The next dose of TRUQAP should be taken at your usual time.
Follow the instructions provided and use TRUQAP until your doctor tells you to stop.
TRUQAP is used in combination with another medicine called fulvestrant which is given as an injection. The recommended dose of fulvestrant is 500 mg administered on Days 1, 15, and 29, and once monthly thereafter.
During your treatment with TRUQAP, for women who have not reached menopause, your doctor will prescribe a medicine called a luteinising hormone releasing hormone (LHRH) agonist.
When to take TRUQAP
TRUQAP should be used at about the same time in the morning and evening of the dosing days.
If you forget to use TRUQAP
If you miss your dose at the usual time, you may still take it within 4 hours from the time you usually take it. If it has been more than 4 hours after you usually take your dose, skip that dose. Take the next dose at your usual time.
Do not take a double dose to make up for the dose you missed.
If you use too much TRUQAP
If you think that you have used too much TRUQAP, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using TRUQAP?
Things you should do
Call your doctor straight away if you experience the following during treatment with TRUQAP:
- Signs of increased blood sugar levels (hyperglycaemia); increased thirst and dry mouth, passing urine more often than usual, increased appetite with weight loss. It is important that you have your blood sugar monitored during treatment.
High blood sugar levels may cause a serious condition called diabetic ketoacidosis, which may become life-threatening if it is not treated at an early stage. Symptoms of diabetic ketoacidosis may include feeling nausea, vomiting, abdominal pain, difficulty breathing, sick or being sick, shortness of breath, severe thirst, feeling weak and unusual tiredness or sleepiness, confusion, a sweet smell to your breath, a sweet or metallic taste in your mouth, a strange odour to your urine or sweat and frequent urination. - Signs of diarrhoea which may include loose or watery stool. Severe diarrhoea may cause dehydration with symptoms such as dry sticky mouth, severe thirst, dizziness, fatigue, dark-coloured urine or urinating less often than normal or not at all.
- Rash, reddening of the skin, blistering of the lips, eyes or mouth, skin peeling, dry skin, skin inflammation with rash, shedding and/or scaling of skin surface.
Your doctor may need to treat these symptoms, temporarily interrupt your treatment, reduce your dose, or permanently stop your treatment with TRUQAP.
Remind any doctor, dentist or pharmacist you visit that you are using TRUQAP.
Ensure you are using effective contraception. Even if you stop treatment, you should continue to use effective contraception for at least a further 4 weeks if you are a woman and a further 16 weeks if you are a man.
If you become pregnant while you are taking this medicine or within 4 weeks of stopping this medicine, tell your doctor immediately.
If you are a man and your partner becomes pregnant while you are taking this medicine or within 16 weeks of stopping this medicine, tell your doctor immediately.
Keep all of your doctor's appointments so that your progress can be checked.
Your doctor will do some tests from time to time to make sure the medicine is working and to prevent unwanted side effects. For example, periodic tests will be done to check your blood sugar levels during treatment.
Things you should not do
- Do not stop using this medicine or change the dose unless without checking with your doctor.
- Do not use TRUQAP to treat any other complaints unless your doctor tells you to.
- Do not give your medicine to anyone else, even if they have the same condition as you.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how TRUQAP affects you.
TRUQAP may affect your ability to drive or use machines. If you feel tired while taking TRUQAP, take special care when driving or using tools or machines.
Looking after your medicine
Keep your tablets below 30°C.
Follow the instructions in the carton on how to take care of your medicine properly.
Store your tablets in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Keep it where young children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Some other serious side effects may only become known through tests (for example, low potassium level in the blood). Your doctor will test your blood regularly while you are being treated with TRUQAP.
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What TRUQAP contains
| Active ingredient (main ingredient) | capivasertib |
| Other ingredients (inactive ingredients) | microcrystalline cellulose calcium hydrogen phosphate croscarmellose sodium magnesium stearate hypromellose titanium dioxide macrogol 3350 polydextrose copovidone medium chain triglycerides iron oxide black iron oxide red iron oxide yellow |
Do not take this medicine if you are allergic to any of these ingredients.
What TRUQAP looks like
TRUQAP 160 mg tablets are round, biconvex, beige film-coated tablets debossed with ‘CAV’ above ‘160’ on one side and plain on the reverse. (Aust R 407961)
TRUQAP 200 mg tablets are capsule-shaped, biconvex, beige film-coated tablets debossed with ‘CAV 200’ on one side and plain on the reverse. (Aust R 407960).
Who distributes TRUQAP
AstraZeneca Pty Ltd
ABN 54 009 682 311
66 Talavera Road
MACQUARIE PARK NSW 2113
Telephone: 1800 805 342
This leaflet was prepared in September 2025.
TRUQAP is a registered trade mark of the AstraZeneca group of companies
© AstraZeneca, 2025
VV-RIM-06054714 v5.0
Brand Information
| Brand name | Truqap |
| Active ingredient | Capivasertib |
| Schedule | S4 |
▼ This medicinal product is subject to additional monitoring in Australia. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse events at www.tga.gov.au/reporting-problems.
MIMS Revision Date: 01 October 2025
1 Name of Medicine
Capivasertib.
2 Qualitative and Quantitative Composition
Truqap 160 mg. Each film-coated tablet contains 160 mg of capivasertib.
Truqap 200 mg. Each film-coated tablet contains 200 mg of capivasertib.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Film-coated tablet.
Truqap 160 mg tablets are round, biconvex, beige film-coated tablets debossed with 'CAV' above '160' on one side and plain on the reverse.
Truqap 200 mg tablets are capsule-shaped, biconvex, beige film-coated tablets debossed with 'CAV 200' on one side and plain on the reverse.
4 Clinical Particulars
4.1 Therapeutic Indications
Truqap is indicated in combination with fulvestrant for the treatment of adult patients with hormone receptor (HR) positive, human epidermal growth factor receptor 2 (HER2) negative (defined as IHC 0 or 1+, or IHC 2+/ISH-) locally advanced or metastatic breast cancer following recurrence or progression on or after an endocrine based regimen.
4.2 Dose and Method of Administration
Treatment with Truqap should be initiated and supervised by a physician experienced in the use of anticancer medicinal products.
The recommended dose of Truqap in combination with fulvestrant is 400 mg (two 200 mg tablets) taken orally twice daily approximately 12 hours apart (total daily dose of 800 mg) with or without food, for 4 days followed by 3 days off treatment. See Table 1.
The recommended dose of fulvestrant is 500 mg administered on Days 1, 15, and 29, and once monthly thereafter. Refer to the approved Product Information of fulvestrant for more information.
In pre/peri-menopausal women, Truqap plus fulvestrant should be combined with a luteinising hormone releasing hormone (LHRH) agonist according to current clinical practice standards.
For men, consider administering a LHRH agonist according to current clinical practice standards.
If a dose of Truqap is missed, it can be taken within 4 hours after the time it is usually taken. After more than 4 hours, the dose should be skipped. The next dose of Truqap should be taken at the usual time. There should be at least 8 hours between doses. If the patient vomits, an additional dose should not be taken. The next dose of Truqap should be taken at the usual time.
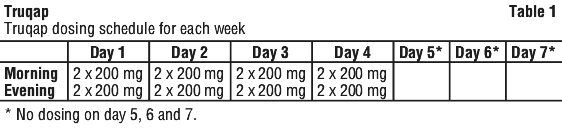
Dose adjustments. For adverse reactions. Treatment with Truqap may be interrupted to manage adverse reactions and dose reduction can be considered. Dose reductions for capivasertib should be carried out as described in Table 2. The dose of capivasertib can be reduced up to two times. Dose modification guidance for specific adverse reactions is presented in Table 3-5.




Co-administration with moderate CYP3A4 inhibitors. When concomitantly used with a moderate CYP3A inhibitor, reduce the dosage of Truqap to 320 mg twice daily (equivalent to a total dose of 640 mg) and monitor patients for adverse reactions (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Special patient populations. Renal impairment. No dose adjustment is required for patients with mild or moderate renal impairment. Truqap is not recommended for patients with severe renal impairment, as safety and pharmacokinetics have not been studied in these patients (see Section 5.2 Pharmacokinetic Properties).
Hepatic impairment. No dose adjustment is required for patients with mild hepatic impairment. Limited data are available for patients with moderate hepatic impairment; Truqap should be administered to patients with moderate hepatic impairment only if the benefit outweighs the risk and these patients should be monitored closely for signs of toxicity. Truqap is not recommended for patients with severe hepatic impairment, as safety and pharmacokinetics have not been studied in these patients (see Section 5.2 Pharmacokinetic Properties).
Use in the elderly. No dose adjustment is required for elderly patients (see Section 5.2 Pharmacokinetic Properties). There are limited data in patients aged ≥ 75 years.
Paediatric use. Truqap is not indicated for use in paediatric patients, as safety and efficacy of Truqap in children and adolescents have not been established.
Method of administration. Truqap tablets should be swallowed whole with water and not chewed, crushed dissolved, or divided. No tablets should be ingested if it is broken, cracked, or otherwise not intact.
4.3 Contraindications
Prior severe hypersensitivity to the active substance or to any of the excipients listed, see Section 6.1 List of Excipients.
4.4 Special Warnings and Precautions for Use
Hyperglycaemia. Severe hyperglycaemia has occurred in 8 (2.3%) patients treated with Truqap. Severe hyperglycaemia, associated with diabetic ketoacidosis (DKA) and ketoacidosis, was reported in patients treated with Truqap, including in one patient treated with Truqap in CAPItello-291 (see Section 4.8 Adverse Effects (Undesirable Effects)). Some cases of DKA have been reported with fatal outcomes. DKA can occur at any time during Truqap treatment. In some reported cases, DKA developed in less than 10 days.
Before initiating treatment with Truqap, inform patients about Truqap's potential to cause hyperglycaemia and request for them to immediately contact their healthcare professional if hyperglycaemia symptoms (e.g. excessive thirst, urinating more often than usual or greater amount of urine than usual, increased appetite with weight loss) occur.
In a setting of additional co-morbidities (e.g. dehydration, malnourishment, concurrent chemotherapy/steroids, sepsis and cancer) the risk of hyperglycaemia progressing to diabetic ketoacidosis may be higher. DKA should be considered as one of the differential diagnoses in the event of additional non-specific symptoms such as nausea, vomiting, abdominal pain, difficulty breathing, fruity odour on breath, confusion, unusual fatigue, or sleepiness. In patients where DKA is suspected, Truqap treatment should be interrupted immediately. If DKA is confirmed, then Truqap should be permanently discontinued.
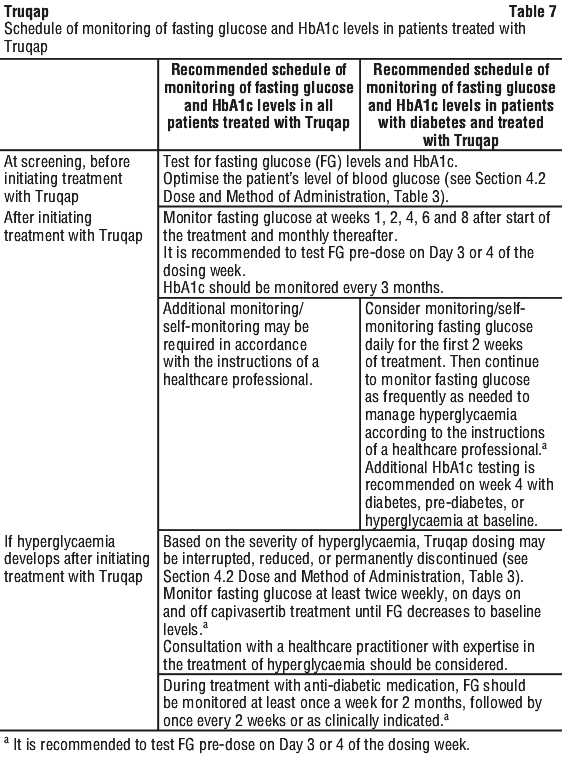
Patients must be tested for fasting blood glucose (FG) levels and HbA1C prior to start of treatment with Truqap and in accordance with the intervals stated in Table 7. Based on the severity of hyperglycaemia, Truqap dosing may be interrupted, reduced, or permanently discontinued (see Section 4.2 Dose and Method of Administration, Table 3).
More frequent blood glucose monitoring is recommended in patients that develop hyperglycaemia during treatment, those with baseline risk factors for DKA (including but not exclusive to diabetes mellitus, pre-diabetes, those receiving regular oral steroids) and in those that develop risk factors for DKA during treatment (e.g. infection, sepsis, raised HbA1c) (see Table 7). In addition to FG, monitoring of ketones (preferably in blood) and other metabolic parameters (as indicated) is recommended when a patient experiences hyperglycaemia.
In addition to the recommended management of hyperglycaemia described, see Section 4.2 Dose and Method of Administration, Table 3, counselling on lifestyle changes is recommended for patients with baseline risk factors and those that develop hyperglycaemia during treatment with Truqap.
The safety of Truqap in patients with Type 1 and Type 2 diabetes requiring insulin has not been studied as these patients were excluded from clinical study. Patients with history of diabetes mellitus may require intensified diabetic treatment and should be closely monitored. See Table 7.
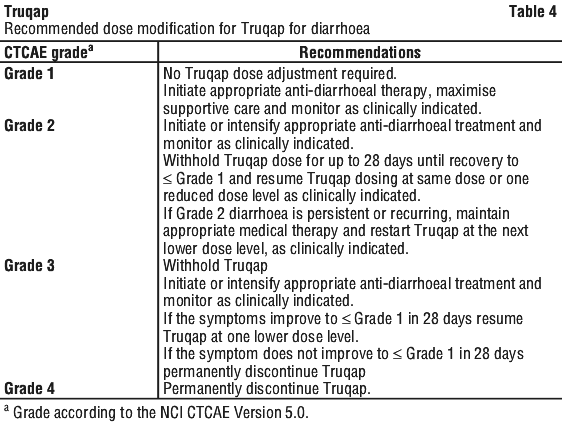
Diarrhoea has been frequently reported in patients treated with Truqap (see Section 4.8 Adverse Effects (Undesirable Effects)).
Based on the severity of diarrhoea, Truqap dosing may be interrupted, reduced or permanently discontinued (see Section 4.2 Dose and Method of Administration, Table 4). Advise patients to start anti-diarrhoeal treatment at the first sign of diarrhoea, increase oral fluids if diarrhoea symptoms occur while taking Truqap. Maintenance of normovolaemia and electrolyte balance is required in patients with diarrhoea to avoid complications related to hypovolaemia and low electrolyte levels.
Rash and other skin reactions. Skin reactions, including erythema multiforme and dermatitis exfoliative generalised, were reported in patients receiving Truqap. Drug reaction with eosinophilia and systemic symptoms (DRESS) was reported in one patient treated with Truqap in CAPItello-291. Palmar-plantar erythrodysesthesia was reported in 3 patients treated with Truqap in CAPItello-291.
Patients should be monitored for signs and symptoms of rash or dermatitis and based on severity of skin drug reactions the dosing may be interrupted, reduced, or permanently discontinued (see Section 4.2 Dose and Method of Administration, Table 5). Early consultation with a dermatologist is recommended to ensure greater diagnostic accuracy and appropriate management.
Patients excluded from the study. The patients with history of clinically significant cardiac disease including QTcF > 470 msec, any factors that increased the risk of QTc prolongation or risk of arrhythmic events or risk of cardiac function impairment were excluded from CAPItello 291. This should be considered if Truqap is prescribed in these patients.
Use in the elderly. No dose adjustment is required for elderly patients (see Section 5.2 Pharmacokinetic Properties). There are limited data in patients aged ≥ 75 years.
Paediatric use. The safety and efficacy of Truqap in children aged 0-18 years of age has not been established.
Effects on laboratory tests. See Section 4.4 Special Warnings and Precautions for Use; Section 4.8 Adverse Effects (Undesirable Effects).
4.5 Interactions with Other Medicines and Other Forms of Interactions
Effects of other medicinal products on capivasertib. In vitro studies have demonstrated that capivasertib is primarily metabolised by CYP3A4 and UGT2B7 enzymes. Capivasertib is a substrate for P-glycoprotein (P-gp) and OCT2.
In a study in healthy subjects, co-administration of multiple 200 mg doses of the strong CYP3A4 inhibitor itraconazole with a single 80 mg capivasertib dose increased capivasertib AUC and Cmax by 95% and 70%, respectively, relative to a single 80 mg capivasertib dose given alone. At the therapeutic dose regimen, the predicted increase in capivasertib AUC and Cmax by itraconazole is between 52% and 56%, and between 30% and 35%, respectively, over a dosing cycle.
In a study in patients with prostate cancer, the strong CYP3A4 inducer enzalutamide decreased the capivasertib AUC by approximately 40% to 50% and rifampicin is predicted to decrease capivasertib AUC by approximately 70%.
Co-administration of a single dose of capivasertib 400 mg after repeated dosing of acid-reducing agent rabeprazole 20 mg twice daily for 3 days in healthy subjects did not result in clinically relevant changes of the capivasertib exposure. The capivasertib AUC and Cmax decreased by 6% and 27% respectively when administered with and without rabeprazole. In addition, a population pharmacokinetic analysis showed no significant impact of co-administration of acid reducing agents on the pharmacokinetics of capivasertib in patients. Capivasertib can be taken with acid reducing agents.
Based on physiologically based pharmacokinetic models, the predicted increase in capivasertib AUC by the moderate inhibitors verapamil and erythromycin is approximately 40%, with less impact on Cmax. Co-administration with the UGT2B7 inhibitor probenecid is predicted to cause an increase in capivasertib AUC of 23 to 37% over a dosing cycle.
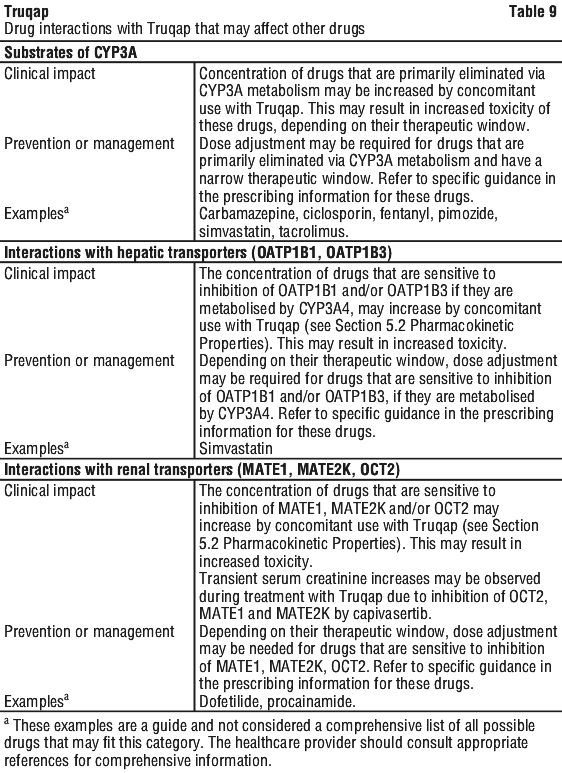
Effect of other drugs on Truqap. See Table 8.
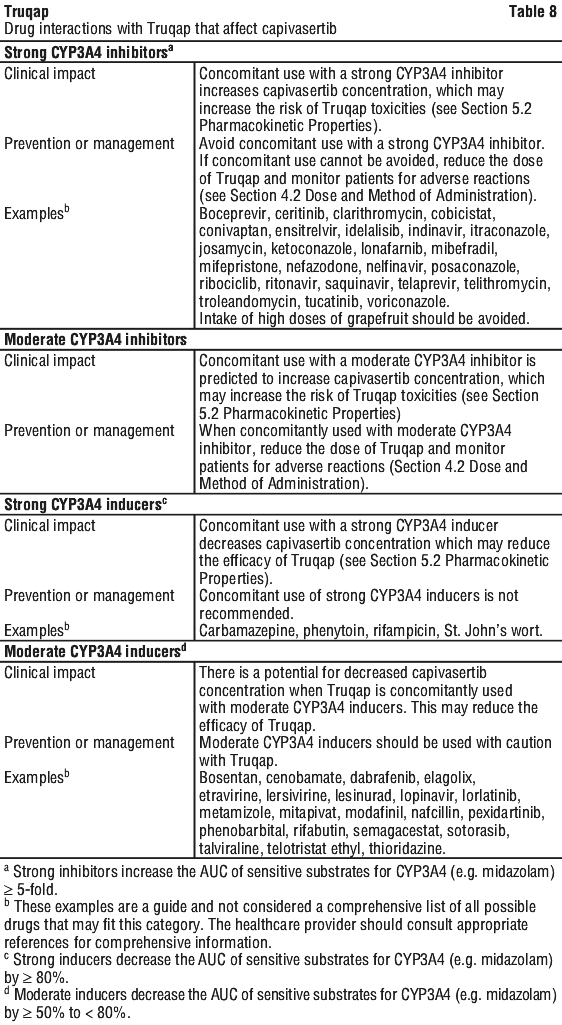
Capivasertib inhibited CYP2C9, CYP2D6, CYP3A4 and UGT1A1 metabolising enzymes and BCRP, OATP1B1, OATP1B3, OAT3, OCT2, MATE1 and MATE2K drug transporters in in vitro studies. Capivasertib induced CYP3A4 in vitro in human hepatocytes.
Based on in vitro data and physiologically based modelling, capivasertib was predicted to have no effect on the AUC of CYP2C9, CYP2D6 or UGT1A1 substrates, atorvastatin or rosuvastatin.
Effect of Truqap on other drugs. See Table 9.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. There are no clinical data on fertility. In repeat-dose toxicity studies, capivasertib resulted in tubular degeneration in the testes and cellular debris and decreased spermatocytes in the epididymides in mice, rats and dogs at oral doses of 150, 100 and 15 mg/kg/day, respectively (exposure similar to the clinical exposure in humans based on AUC). Capivasertib had no effects on fertility in male rats at doses up to 100 mg/kg/day for 10 weeks. The effect on female fertility in animals has not been studied.
Use in pregnancy. (Category D)
There are no data from the use of Truqap in pregnant women. Studies in animals have shown reproductive toxicity. In a rat embryo-fetal study, capivasertib caused an increase in post implantation loss, an increase in early embryonic deaths, together with reduced gravid uterine and fetal weights, and minor fetal visceral variations (left sided umbilical artery). These effects were seen at a dose level of 150 mg/kg/day which caused maternal toxicity, and where plasma concentrations were approximately 0.8 times the exposure in humans at the recommended dose of 400 mg twice daily (based on total AUC). When capivasertib was administered to pregnant rats at 150 mg/kg/day throughout gestation and through early lactation, there was a reduction in litter and pup weights. PI3K/AKT/mTOR signalling plays important roles in embryo-fetal development. A range of growth defects were seen in Akt knockout mice. Therefore, Truqap is not recommended during pregnancy and in women of childbearing potential not using contraception.
Contraception in males and females. Women of childbearing potential should be advised to avoid becoming pregnant while receiving Truqap. A pregnancy test should be performed for women of childbearing potential prior to initiating treatment, and verified as negative prior to initiating treatment, and re-testing considered throughout treatment.
Patients should be advised to use effective contraception during treatment with Truqap and for the following periods after completion of treatment with Truqap: at least 4 weeks for females and 16 weeks for males.
Please see Section 4.6 Fertility, Pregnancy and Lactation of the approved product information for fulvestrant.
Use in lactation. It is not known whether capivasertib or its metabolites are excreted in human milk. Exposure to capivasertib was confirmed in suckling rat pups which may indicate the excretion of capivasertib in milk. A risk to the suckling child cannot be excluded. Breastfeeding should be discontinued during treatment with Truqap.
4.7 Effects on Ability to Drive and Use Machines
During treatment with capivasertib, fatigue has been reported and those patients who experience this symptom should be advised to observe caution when driving or operating machinery.
4.8 Adverse Effects (Undesirable Effects)
Overall summary of the safety profile. Severe hyperglycaemia, diarrhoea, and skin reactions have occurred in patients taking capivasertib (see Section 4.4 Special Warnings and Precautions for Use).
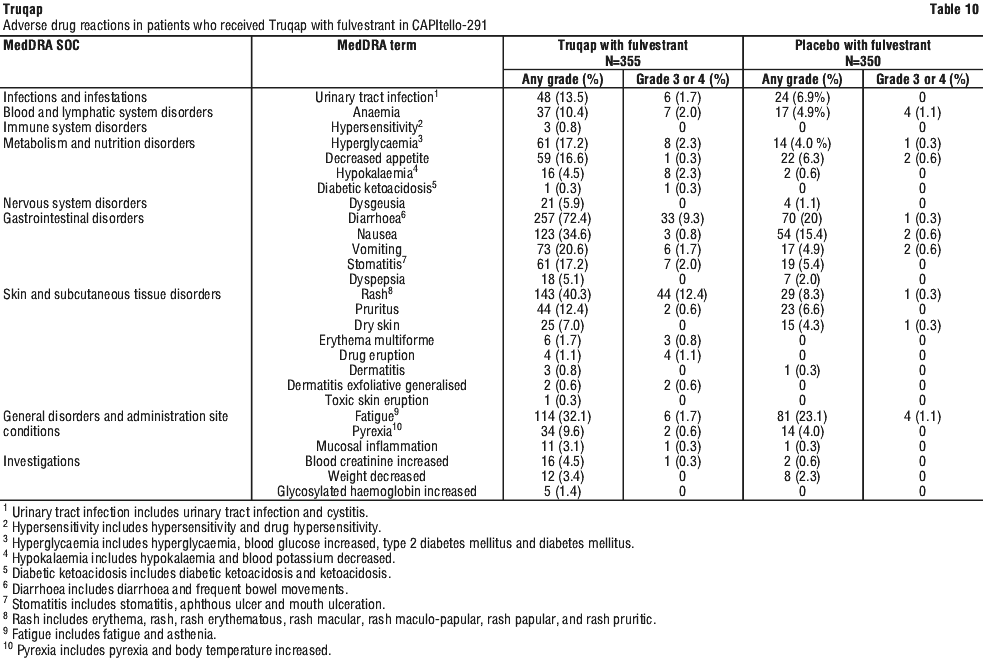
The safety profile of Truqap is based on data from 355 patients who received Truqap plus fulvestrant in CAPItello-291.
The most common adverse reactions (reported at a frequency of ≥ 20%), were diarrhoea (72.4%), rash (40.3%), nausea (34.6%), fatigue (32.1%) and vomiting (20.6%). The most common grade 3 or 4 adverse reactions (reported at frequency ≥ 2%) were rash (12.4%), diarrhoea (9.3%), hyperglycaemia (2.3%), hypokalaemia (2.3%), anaemia (2.0%), and stomatitis (2.0%).
Serious adverse reactions (SARs) were seen in 24 (6.8%) patients receiving Truqap plus fulvestrant. Serious adverse reactions reported in ≥ 1% of patients receiving Truqap plus fulvestrant included rash 8 (2.3%), diarrhoea 6 (1.7%), and vomiting 4 (1.1%).
Dose reductions due to adverse reactions were reported in 63 (17.7%) patients. The most common adverse reactions (reported at frequency ≥ 1%) leading to dose reduction of Truqap were diarrhoea (7.9%) and rash (4.5%).
Treatment discontinuation due to adverse reactions occurred in 35 (9.9%) patients. The most common adverse reactions (reported at frequency ≥ 1%) leading to treatment discontinuation were rash (4.5%), diarrhoea (2.0%), and vomiting (2.0%).
Fatal adverse events regardless of causal association with Truqap occurred in 4 (1.1%) of patients who received Truqap with fulvestrant, including 1 (0.3%) pneumonia aspiration, 1 (0.3%) sepsis, 1 (0.3%) cerebral haemorrhage and 1 (0.3%) acute myocardial infarction.
Adverse drug reactions. Adverse drug reactions (ADRs) are organised by MedDRA System Organ Class (SOC). Within each SOC, preferred terms are arranged by decreasing frequency and then by decreasing seriousness. Frequencies of occurrence of adverse reactions are defined as: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1000); very rare (< 1/10,000) and not known (cannot be estimated from available data). See Table 10.
Of the 61 patients with hyperglycaemia, in 38 (62.3%) patients hyperglycaemia improved or resolved at treatment discontinuation or last follow up.
Diarrhoea. Diarrhoea occurred in 257 (72.4%) patients receiving Truqap. Grade 3 and/or 4 diarrhoea occurred in 33 (9.3%) patients. Dose reduction was required in 28 (7.9%) patients and 7 (2.0%) patients discontinued Truqap due to diarrhoea. In the 257 patients with diarrhoea, anti-diarrhoeal medication was required in 59% (151/257) of patients to manage diarrhoea symptoms.
Rash. Rash (including erythema, rash, rash erythematous, rash macular, rash maculo-papular, rash papular, and rash pruritic) was reported in 143 (40.3%) patients. Grade 3 and/or 4 occurred in 44 (12.4%) of patients who received capivasertib. Dose reduction was required in 16 (4.5%) patients and 16 (4.5%) patients discontinued Truqap due to rash.
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
4.9 Overdose
There is currently no specific treatment in the event of an overdose with Truqap and possible symptoms of overdose are not established. Physicians should follow general supportive measures and patients should be treated symptomatically.
For information on the management of overdose, contact the Poison Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action. Capivasertib is an inhibitor of the kinase activity of all 3 isoforms of serine/threonine kinase AKT (AKT1, AKT2 and AKT3). AKT is a pivotal node in the phosphatidylinositol 3-kinase (PI3K) signalling cascade regulating multiple cellular processes including cellular survival, proliferation, cell cycle, metabolism, gene transcription and cell migration. AKT activation in tumours is a result of upstream activation from other signalling pathways, mutations of AKT, loss of Phosphatase and Tensin Homolog (PTEN) function and mutations in the catalytic subunit of PI3K (PIK3CA).
Capivasertib inhibits the phosphorylation of AKT substrates such as glycogen synthase kinase 3-β (GSK3β) and proline-rich AKT substrate of 40 kilodaltons (PRAS40). Capivasertib reduces growth of a range of cell lines derived from solid tumours and haematological disease. Multiple breast cancer cell lines were sensitive to capivasertib monotherapy. Within cell lines showing greater sensitivity to capivasertib there was an enrichment of PIK3CA or AKT1 mutations, or loss of PTEN. Some cell lines lacking such mutations were also sensitive to capivasertib.
In vivo, monotherapy, capivasertib inhibits growth of human cancer xenograft models representative of different tumour types. including ER+ and triple negative breast cancer models with PIK3CA, AKT1 mutations, PTEN loss and HER2 amplification, mutant xenograft models and triple negative breast cancer xenograft models. Combined treatment with capivasertib and fulvestrant demonstrated a greater anti-tumour response in a range of human breast cancer PDX models representative of different breast cancer subsets. This included models without detectable mutations or alterations in PIK3CA, PTEN or AKT, as well as models with mutations or alterations in PIK3CA, PTEN or AKT.
Cardiac electrophysiology. Based on an exposure-response modelling analysis of data from 180 patients with advanced solid malignancies who received capivasertib doses from 80 to 800 mg a linear relationship between capivasertib concentration and increases in the QTcF interval was projected. The estimated QTcF prolongation was 3.87 ms at the steady state Cmax following 400 mg twice daily and the exposure that is predicted to cause a QTcF prolongation of 20 ms is approximately 4- to 5-fold higher than the therapeutic Cmax. No clinically relevant effect of capivasertib on QT prolongation associated with pro-arrhythmic effect was observed at the recommended dose of 400 mg twice daily in the pivotal study. Patients with QTcF > 470 were not included in CAPItello-291.
Clinical trials. CAPItello-291 was a randomised, double-blind, placebo-controlled study designed to demonstrate the efficacy and safety of Truqap in combination with fulvestrant in adult females, pre- or post-menopausal, and adult males with locally advanced (inoperable) or metastatic HR positive and HER2 negative breast cancer following recurrence or progression on or after aromatase inhibitor (AI) based treatment. The trial enrolled 708 adult patients with locally advanced (inoperable) or metastatic HR-positive, HER2-negative breast cancer irrespective of PIK3CA/AKT1/PTEN-alteration status.
PIK3CA/AKT1/PTEN alteration status was identified centrally and retrospectively by Next Generation Sequencing (NGS) using the FoundationOne CDx assay, excluding in China where a Burning Rock Oncoscreen Plus assay was used, and classified in accordance with the principles published in the American College of Medical Genetics and Genomics (ACMG) standards and guidelines for the interpretation of sequence variants.
Patients with qualifying alterations in PIK3CA/AKT1/PTEN genes were included in the analyses for the overall population and the PIK3CA/AKT1/PTEN altered subgroup. 289 (40.8%) patients, 155 (43.7%) in capivasertib arm and 134 (38.0%) in the placebo arm met these criteria.
A total of 313 patients (44.2%) had tumours that did not harbour qualifying mutations in the PIK3CA/AKT1/PTEN genes. 142 (40.0%) of patients included in the capivasertib treatment arm and 171 (48.4%) in the placebo arm met these criteria.
Of the 106 patients (15% of total study participants) with unknown PIK3CA/AKT1/PTEN alteration status, 14 (13%) were unknown due to no sample availability, 73 (69%) because of preanalytical failure, and 19 (18%) because of post analytical failure.
Patients were to have received treatment with an AI-containing regimen (single agent or in combination) and have:
Radiological evidence of breast cancer recurrence or progression while on, or within 12 months of the end of (neo)adjuvant treatment with an AI, or
Radiological evidence of progression while on prior AI administered as a treatment line for locally advanced or metastatic breast cancer (this did not need to be the most recent therapy).
Patients were excluded if they had more than 2 lines of endocrine therapy for locally advanced (inoperable) or metastatic disease, more than 1 line of chemotherapy for locally advanced (inoperable) or metastatic disease, prior treatment with AKT, PI3K, mTOR inhibitors, fulvestrant and/or other SERDs, clinically significant abnormalities of glucose metabolism (defined as patients with diabetes mellitus Type 1 or Type 2 requiring insulin treatment, and/or HbA1c ≥ 8.0% (63.9 mmol/mol)), history of clinically significant cardiac disease, and symptomatic visceral disease or any disease burden that makes the patient ineligible for endocrine therapy.
A total of 708 patients were randomised 1:1 to receive either 400 mg of Truqap (N=355) or placebo (N=353) given twice daily for 4 days followed by 3 days off treatment each week of 28-day treatment cycle. Fulvestrant 500 mg was administered on cycle 1 days 1 and 15 and then at day 1 of a 28-day cycle. Peri/pre-menopausal women were treated with an LHRH agonist. Randomisation was stratified by presence of liver metastases, prior treatment with CDK4/6 inhibitors and geographical region (region 1: US, Canada, Western Europe, Australia and Israel vs region 2: Latin America, Eastern Europe and Russia vs Region 3: Asia). Treatment was administered until disease progression, death, withdrawal of consent, or unacceptable toxicity. A tumour sample was collected prior to randomisation to determine PIK3CA/AKT1/PTEN alteration status retrospectively by central testing.
Demographic and baseline characteristics were well balanced between arms. Of the 708 patients, the median age was 58 years (range 26 to 90); female (99%); White (57.5%), Asian (26.7%), Black (1.1%); Eastern Cooperative Oncology Group (ECOG) performance status 0 (65.7%), 1 (34.2%), 21.8% were pre/peri menopausal. All patients received prior endocrine-based therapy (100% AI based treatment and 44.1% received tamoxifen). Prior treatment with CDK4/6 inhibitor was reported in 70.1% of patients. Chemotherapy for locally advanced (inoperable) or metastatic disease was reported in 18.2% of patients. Patient demographics for those in the PIK3CA/AKT1/PTEN-altered subgroup were generally representative of the overall study population.
The dual primary endpoints were investigator assessed progression free survival (PFS) in the overall population and PFS in the PIK3CA/AKT1/PTEN-altered subgroup per Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. The key secondary endpoints of overall survival (OS) and objective response rate (ORR) will be formally analysed at future data cut offs.
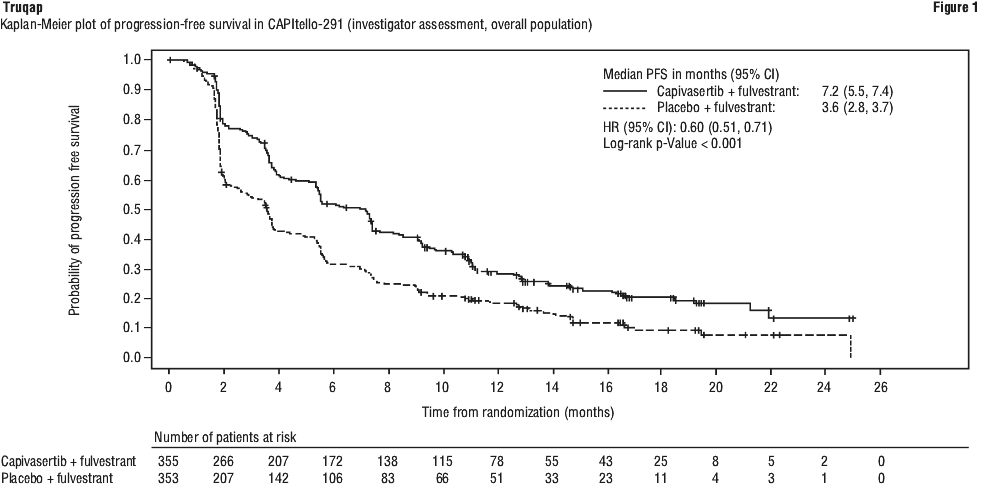
At the time of primary analysis, the median duration of follow-up for PFS in the overall population was 13 months (range: 0 to 25 months) in censored patients.
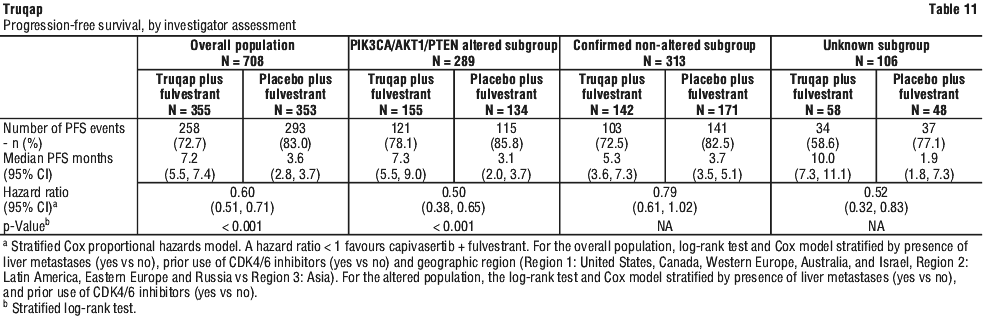
The study demonstrated statistically significant and clinically meaningful improvement in PFS for patients receiving Truqap plus fulvestrant compared to patients receiving placebo plus fulvestrant, in both the overall population and the PIK3CA/AKT1/PTEN-altered subgroup. An exploratory analysis of PFS in the 313 (44%) patients in the Known Non-altered Population (no PIK3CA/AKT1/PTEN altered tumour) showed a HR (95% CI) of 0.79 (0.61 to 1.02). The median PFS (95% CI) was 5.3 months (3.6 to 7.3) in the capivasertib group compared with 3.7 months (3.5 to 5.1) in the placebo group. An exploratory analysis of PFS in the 106 (15%) patients in the No Result Population showed a HR (95% CI) of 0.52 (0.32 to 0.83). The median PFS (95% CI) was 10.0 months (7.3 to 11.1) in the capivasertib group compared with 1.9 months (1.8 to 7.3) in the placebo group. PFS results by investigator assessment were supported by consistent results from a blinded independent review committee (BIRC) assessment. Scans were repeated every 8 weeks (± 7 days) for the first 18 months and every 12 weeks (± 7 days) thereafter, after start of treatment until objective radiological disease progression as defined by RECIST v1.1.
A preliminary assessment of OS in the overall population (28% maturity) and altered population (30% maturity) at the time of the primary PFS analysis does not suggest a detrimental effect on survival of treatment with capivasertib plus fulvestrant compared with placebo plus fulvestrant. The investigator-assessed ORR in patients receiving Truqap plus fulvestrant and placebo plus fulvestrant was 22.9% and 12.2%, respectively, in the overall population and 28.8% and 9.7%, respectively, in the altered subgroup. The investigator-assessed ORR in patients receiving capivasertib plus fulvestrant and placebo plus fulvestrant was 17.1% and 14.5%, respectively, in the Known Non-altered Population and 22.4% and 11.4%, respectively, in the unknown subgroup.
Efficacy results for overall population and PIK3CA/AKT1/PTEN-altered subgroup are presented in Table 11 and Figure 1 and Figure 2.
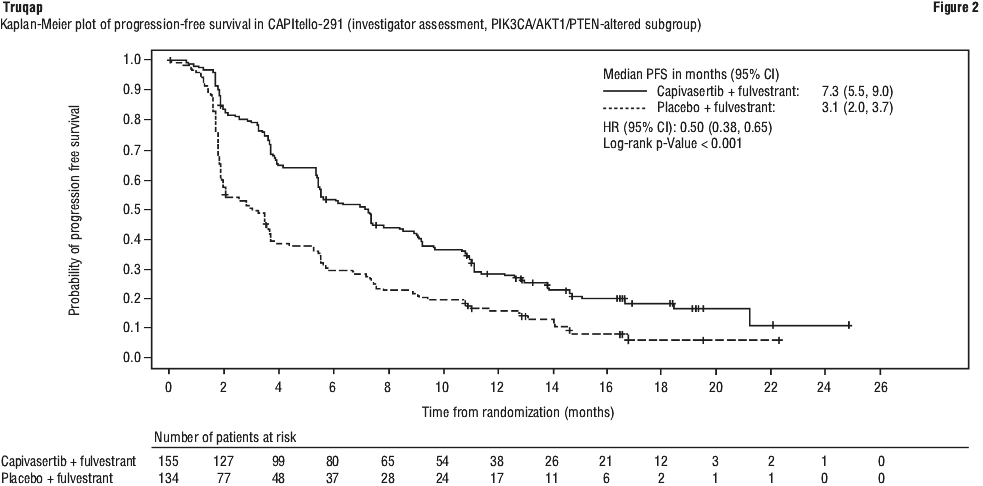
5.2 Pharmacokinetic Properties
Capivasertib pharmacokinetics have been characterised in healthy subjects and patients with solid tumours. The systemic exposure (AUC and Cmax) increased approximately proportionally to the dose over the 80 to 800 mg dose range when given to patients. Following intermittent dosing of capivasertib 400 mg twice daily, 4 days on, 3 days off, steady-state levels are predicted to be attained on every 3rd and 4th dosing day each week, starting from week 2. During the off-dosing days, the plasma concentrations are low (approximately 0.5% to 15% of the steady state Cmax).
Absorption. Capivasertib is rapidly absorbed with peak concentration (Cmax) observed at approximately 1-2 hours in patients. The mean absolute bioavailability is 29%.
Food effect. When capivasertib was administered after a high-fat, high-calorie meal (approximately 1000 kcal), the fed to fasted ratio was 1.32 and 1.23, for AUC and Cmax, respectively, compared to when given after an overnight fast. When capivasertib was administered after a low-fat, low-calorie (approximately 400 kcal), the exposure was similar to that after fasted administration with fed to fasted ratios of 1.14 and 1.21, for AUC and Cmax, respectively. Co-administration with food did not result in clinically relevant changes to the exposure.
Distribution. The mean volume of distribution (Vss) was 205 L after intravenous administration to healthy subjects. Capivasertib is not extensively bound to plasma protein (percentage unbound 22%) and the plasma to blood ratio is 0.71.
Metabolism. Capivasertib is primarily metabolised by CYP3A4 and UGT2B7 enzymes. The major metabolite in human plasma was an ether glucuronide that accounted for 83% of total drug-related material. A minor oxidative metabolite was quantified at 2% and capivasertib accounted for 15% of total circulating drug-related material. No active metabolites have been identified.
Excretion. The effective half-life after multiple dosing in patients was 8.3 hours. The mean total plasma clearance was 38 L/h after a single intravenous administration to healthy subjects. The mean total oral plasma clearance was 60 L/h after single oral administration and decreased by 8% after repeated dosing of 400 mg twice daily.
Following single oral dose of 400 mg, the mean total recovery of radioactive dose was 45% from urine and 50% from faeces. Renal clearance was 21% of total clearance. Capivasertib is primarily eliminated by metabolism.
Special populations. Effect of race, age, gender and weight. There were no clinically significant differences in pharmacokinetics of capivasertib based on race/ethnicity (including White and Asian patients), gender or age. There was a statistically significant correlation of apparent oral clearance of capivasertib to body weight. Compared to a patient with a body weight of 66 kg, a 47 kg patient is predicted to have 12% higher AUC. There is no basis for dose modification based on body weight as the predicted effect on capivasertib exposure was small.
Renal impairment. Based on population pharmacokinetic analyses, AUC and Cmax were 1% higher in patients with mild renal impairment (creatinine clearance 60 to 89 mL/min), compared to patients with normal renal function. AUC and Cmax were 16% higher in patients with moderate renal impairment (creatinine clearance 30 to 59 mL/min), compared to patients with normal renal function.
There is no data in severe renal impairment or end-stage renal disease (creatinine clearance < 30 mL/min).
Hepatic impairment. Based on population pharmacokinetic analyses, AUC and Cmax were 5% higher in patients with mild hepatic impairment (bilirubin ≤ ULN and AST > ULN, or bilirubin > 1 ULN to ≤ 1.5 ULN), compared to patients with normal hepatic function. No dose adjustment is required for patients with mild hepatic impairment.
Based on limited data the AUC and Cmax was 17% and 13% higher respectively in patients with moderate hepatic impairment (bilirubin > 1.5 ULN to ≤ 3 ULN), compared to patients with normal hepatic function. There is limited data in patients with moderate hepatic impairment and no data in severe hepatic impairment.
5.3 Preclinical Safety Data
Genotoxicity. Capivasertib showed no mutagenic or genotoxic potential in vitro. Capivasertib was not mutagenic in a bacterial reverse mutation (Ames) assay, or genotoxic in mouse lymphoma cells in vitro. In rats treated orally with doses of 150 mg/kg/day, capivasertib induced micronuclei in the bone marrow via an aneugenic mode of action. These findings occurred at plasma concentrations similar to those observed in humans at the maximum recommended clinical dose of 400 mg twice daily (based on Cmax or AUC).
Carcinogenicity. In a 2-year rat carcinogenicity study there was an increased incidence and/or severity of islet of Langerhans hypertrophy/ hyperplasia (males and females) and neoplastic findings in the testis in males. Findings were observed at exposures lower than those in humans (0.20-0.41 times) at the recommended dose of 400 mg twice daily (based on total AUC).
Non-clinical/repeat-dose toxicity. The major target organs or systems for toxicity were insulin signalling (increased levels of glucose and insulin in mice, rats and dogs), the male reproductive organs (tubular degeneration in mice, rats and dogs), and the renal system in rats (polyuria, decreased tubular epithelial cell size, decreased kidney size and weight). The findings present following 1 month of dosing were largely reversible within 1 month of cessation of dosing. Findings occurred at plasma concentrations lower or similar to those in humans (approximately 0.14 to 2 times) at the recommended dose of 400 mg twice daily (based on total AUC).
Lens degeneration was observed in male rats in the 2-year rat carcinogenicity study at exposures lower than those in humans (0.08 times) at the recommended dose of 400 mg twice daily (based on total AUC) and may be related to elevated glucose levels.
6 Pharmaceutical Particulars
6.1 List of Excipients
Tablet core. Microcrystalline cellulose, calcium hydrogen phosphate, croscarmellose sodium, magnesium stearate.
Tablet coating. Hypromellose, titanium dioxide, macrogol 3350, polydextrose, copovidone, medium chain triglycerides, iron oxide black, iron oxide red, iron oxide yellow.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C.
6.5 Nature and Contents of Container
Capivasertib 160 mg and 200 mg film-coated tablets are supplied in aluminium/aluminium blisters containing 16 tablets. Each pack contains 64 tablets (4 blisters).
*Not all presentations may be available in Australia.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
7 Medicine Schedule (Poisons Standard)
Prescription only medicine (Schedule 4).
Date of First Approval
09 May 2024
Date of Revision
09 September 2025
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.