Ultomiris 100 mg per mL
Brand Information
| Brand name | Ultomiris 100 mg per mL |
| Active ingredient | Ravulizumab |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Ultomiris 100 mg per mL.
Summary CMI
Ultomiris®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
▼ This medicine is subject to additional monitoring due to approval of an extension of indications. This will allow quick identification of new safety information. You can help by reporting any side effects you may get. You can report side effects to your doctor, or directly at www.tga.gov.au/reporting-problems. See the full CMI for further details.
WARNING: Important safety information is provided in a boxed warning in the full CMI. Read before using this medicine.
1. Why am I using ULTOMIRIS?
ULTOMIRIS contains the active ingredient ravulizumab rch.
For more information, see Section 1. Why am I using ULTOMIRIS? in the full CMI.
2. What should I know before I use ULTOMIRIS?
ULTOMIRIS is a medicine that affects your body's defence system and so can lower the ability of your immune system to fight infections. Using this medicine increases your risk of severe infection and sepsis (infection of the blood). You must be vaccinated against meningococcal infection and be aware of the signs and symptoms of a meningococcal infection. You should seek medical care immediately if you see any signs or symptoms of meningococcal infection. Your doctor will provide you with a 'Patient Safety Card'. You must always carry a 'Patient Safety Card' with you.
Talk to your doctor if you have an infection or have had an allergic reaction to any of the ingredients listed at the end of the CMI.
For more information, see Section 2. What should I know before I use ULTOMIRIS? in the full CMI.
3. What if I am taking other medicines?
Talk to your doctor if you have any other medical conditions or are taking any other medicines. The effect of using ULTOMIRIS on other medicines has not been studied.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use ULTOMIRIS?
- ULTOMIRIS will be given to you directly into the vein (intravenously) by a doctor or nurse. The infusion will take approximately 45 minutes but might take longer according to your body weight.
More instructions can be found in Section 4. How do I use ULTOMIRIS? in the full CMI.
5. What should I know while using ULTOMIRIS?
| Things you should do |
|
| Things you should not do |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using ULTOMIRIS? in the full CMI.
6. Are there any side effects?
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
▼ This medicine is subject to additional monitoring due to approval of an extension of indications. This will allow quick identification of new safety information. You can help by reporting any side effects you may get. You can report side effects to your doctor, or directly at www.tga.gov.au/reporting-problems.
WARNING: SERIOUS MENINGOCOCCAL INFECTIONS
As ULTOMIRIS blocks a part of your immune system it increases the risk of severe infection and sepsis, especially by a type of bacteria called Neisseria meningitidis. This can cause cases of meningitis, which is a major brain inflammation, or a severe infection of the blood.
These infections require urgent and appropriate care as they may become rapidly serious or fatal, or lead to major disabilities. It is important to understand the precautions to take to reduce the risk of these infections and what to do if you are worried you may have an infection (see “What I should know before I use ULTOMIRIS” below or refer to your Patient Safety Card).
- You must be vaccinated against meningococcal infection before starting ULTOMIRIS.
- If you initiate ULTOMIRIS treatment less than 2 weeks after receiving a meningococcal vaccine you must take antibiotics until 2 weeks after you have been vaccinated to reduce the risk of infection with Neisseria meningitidis.
- You need to be aware of the signs and symptoms of meningococcal infection (see “What I should know before I use ULTOMIRIS” below or refer to your Patient Safety Card) and notify your doctor immediately if any of the symptoms occur.
- If you cannot reach your doctor, go to the Emergency Department at your nearest hospital. Show your Patient Safety Card to any doctor or nurse who treats you.
Ultomiris® (uhl-toe-mi-riss)
Active ingredient(s): ravulizumab rch [100 mg per 1mL]
Consumer Medicine Information (CMI)
This leaflet provides important information about using ULTOMIRIS.
You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using ULTOMIRIS.
Where to find information in this leaflet:
1. Why am I using ULTOMIRIS?
2. What should I know before I use ULTOMIRIS?
3. What if I am taking other medicines?
4. How do I use ULTOMIRIS?
5. What should I know while using ULTOMIRIS?
6. Are there any side effects?
7. Product details
1. Why am I using ULTOMIRIS?
ULTOMIRIS contains the active ingredient ravulizumab rch.
ULTOMIRIS belongs to a class of medicines called monoclonal antibodies, that attach to a specific target in the body. Ravulizumab rch has been designed to attach to the C5 complement protein, which is a part of the body's defence system called the ‘complement system’.
ULTOMIRIS is used to treat adults and children with a disease called Paroxysmal Nocturnal Haemoglobinuria (PNH).
ULTOMIRIS is also used to treat adults and children with a disease affecting the blood system and kidneys called atypical haemolytic uraemic syndrome (aHUS). ULTOMIRIS can also be used to treat adult patients with generalised Myasthenia Gravis (gMG), a condition which causes weakness of muscles throughout the body. Patients with gMG can experience challenges with fatigue, drooping eyelids, speech, chewing, movement and breathing, amongst other symptoms.
ULTOMIRIS can also be used to treat adult patients with a disease of the central nervous system that mainly affects the eye nerves and the spinal cord called Neuromyelitis Optica Spectrum Disorder (NMOSD). In NMOSD patients, the eye nerves, brain and spinal cord are attacked and damaged by the immune system working incorrectly, which can lead to loss of sight in one or both eyes, weakness, or loss of movement in the legs or arms, painful spasms, loss of feeling, problems with bladder and bowel function and marked difficulties with activities of daily living.
How it works
In patients with PNH, the complement system is overactive and attacks their red blood cells, which can lead to blood clots, low blood cell counts (anaemia), tiredness, difficulty in functioning, pain, abdominal pain, dark urine, shortness of breath, difficulty swallowing and erectile dysfunction. By attaching to and blocking the C5 protein, ULTOMIRIS can stop complement proteins from attacking red blood cells and control symptoms of the disease such as improving anaemia and tiredness.
In patients with aHUS, their complement system attacks vital organs (such as the kidney), blood cells and blood vessels, which can lead to low blood counts (platelets and red blood cells), reduced or lost kidney function, blood clots, tiredness and difficulty in functioning. ULTOMIRIS can block the body's inflammatory response, and its ability to attack and destroy its own vulnerable blood vessels and control symptoms of the disease including injury to the kidneys.
In gMG patients the immune system (which normally protects the body from infection) attacks itself instead. This autoimmune attack occurs at the connection between nerves and muscles, causing the complement system to damage nerve tissue, resulting in nerves which function poorly.
In patients with NMOSD ULTOMIRIS is presumed to block the body's inflammatory response, and its ability to attack and destroy its own eye nerves, brain and spinal cord, which reduces the risk of relapse or attack of NMOSD. ULTOMIRIS is not intended for acute treatment of NMOSD relapse.
2. What should I know before I use ULTOMIRIS?
Warnings
ULTOMIRIS is a medicine that affects your immune system (your body's defence system). ULTOMIRIS can lower the ability of your immune system to fight infections.
The use of ULTOMIRIS increases your risk of meningococcal infection, a severe infection that can affect the linings of the brain and can spread through the blood and body (sepsis) as well as the risk of other infections (e.g. widespread gonorrhoea).
Due to the importance of rapidly identifying and treating meningococcal infection/sepsis in patients who receive ULTOMIRIS, you will be provided with a 'Patient Safety Card' to carry with you at all times, to ensure you are aware of the following signs and symptoms of a meningococcal infection:
- headache with nausea or vomiting
- headache and a fever
- headache with a stiff neck or stiff back
- fever
- fever and rash
- confusion
- muscle aches with flu-like symptoms
- eyes sensitive to light
Call your doctor immediately and go to the Emergency department at your nearest hospital if you have any of the symptoms listed above.
Patients less than 18 years of age must also be vaccinated against Haemophilus Influenzae and pneumococcal infections.
Ask your doctor for advice about gonorrhoea prevention before using this medicine.
When ULTOMIRIS is given, you may experience reactions to the infusion (drip) (infusion reaction) such as headache, lower back pain, and infusion-related pain. Some patients may experience allergic or hypersensitivity reactions (including anaphylaxis, a serious allergic reaction which causes difficulty breathing or dizziness).
Should you experience an infusion reaction, your doctor will interrupt the infusion of ULTOMIRIS and institute appropriate measures depending on the severity.
Symptoms of an allergic reaction may include:
- shortness of breath, wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not use ULTOMIRIS if:
- you have not been vaccinated against Neisseria meningitidis, a bacteria that causes meningococcal infection, or if it has been less than 2 weeks after receiving your meningococcal vaccination and you are not taking antibiotics to reduce the risk of infection
You should also be aware that vaccination may not prevent this type of infection. You may need antibiotics to prevent infection. You should discuss with your doctor the use of ongoing preventative antibiotics and if they are required. - you have unresolved meningococcal infection
- you are allergic to ravulizumab rch, any other proteins of hamster origin, or any of the ingredients listed at the end of this leaflet.
Always check the ingredients to make sure you can use this medicine.
Check with your doctor if you:
- have any infections, or any other medical conditions
- take any medicines for any other condition
- experience any infusion related pain, when ULTOMIRIS is given, you may experience a reaction to the infusion such as a headache or lower back pain
- are on a controlled sodium diet as ULTOMIRIS contains sodium (main component of cooking/table salt). Each 3mL vial contains 4.6 mg sodium and each 11mL vial contains 16.8 mg sodium. This may need to be considered in calculating your salt/sodium intake.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
ULTOMIRIS has not been studied in pregnant women. Women who may become pregnant should use effective contraception methods during treatment, and for 8 months after stopping treatment.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
It is not known whether ULTOMIRIS passes into breast milk. Since many medicines are secreted into breast milk, breastfeeding should be discontinued during treatment, and for 8 months after stopping treatment.
Elderly
There are no special precautions needed for the treatment of patients aged from 65 years and over, although experience with ULTOMIRIS in elderly patients with PNH and aHUS, or NMOSD in clinical studies is limited.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
The effect of using ULTOMIRIS on other medicines has not been studied.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect ULTOMIRIS.
4. How do I use ULTOMIRIS?
How much to use
- The doses administered are based on your body weight; your doctor will calculate this.
- ULTOMIRIS will be given to you by infusion (drip) into the vein (intravenously) by a doctor or nurse. The infusion will take approximately 45 minutes but might take longer according to your body weight.
- Your first dose is called the loading dose. Two weeks after receiving your loading dose, you will be given a maintenance dose of ULTOMIRIS. This will be repeated every 8 weeks for patients above 20kg, and every 4 weeks for patients weighing less than 20kg.
- If you were previously receiving another medicine for PNH, aHUS or NMOSD called Soliris®, the loading dose should be given 2 weeks after the last SOLIRIS maintenance infusion or 1 week after the last SOLIRIS induction infusion.
If you forget to use ULTOMIRIS
If you forget or miss your appointment for an ULTOMIRIS infusion, please contact your doctor immediately for advice and see Section 5 Things you should not do for information about the risks of interrupting or stopping your ULTOMIRIS treatment.
If you use too much ULTOMIRIS
There have been no reported overdoses of ULTOMIRIS. As ULTOMIRIS is given to you under the supervision of your doctor, it is unlikely that you will receive too much.
5. What should I know while using ULTOMIRIS?
Things you should do
- Always carry your Patient Safety Card with you and show it to any doctor or nurse that treats you.
- Ensure that your meningococcal vaccination is up to date.
- You should also be aware that vaccination may not always prevent this type of infection. Your doctor might consider that you need additional measures, such as antibiotics, to prevent infection.
- Ensure that your child's vaccinations are up to date.
- Be aware of the signs and symptoms of a serious infection. See Warnings under Section 2. What should I know before I use ULTOMIRIS?
Remind any doctor, dentist or pharmacist you visit that you are using ULTOMIRIS.
Things you should not do
- Do not stop using this medicine without checking with your doctor.
Interrupting or stopping treatment with ULTOMIRIS may cause your PNH, aHUS, gMG or NMOSD symptoms to return. Your doctor will discuss the possible side effects with you and explain the risks. - For PNH, the risks of stopping ULTOMIRIS include an increase in the destruction of your red blood cell which may cause:
- a large drop in your red blood cell count causing anaemia. Symptoms of anaemia may include tiredness, headaches, dizziness and looking pale
- dark urine
- fatigue
- abdominal pain
- shortness of breath
- difficulty swallowing
- erectile dysfunction (impotence)
- confusion or a change in how alert you are
- chest pain or angina
- blood clots
- an increase in your serum creatinine level (problems with kidneys).
If you experience any of these symptoms, contact your doctor immediately.
Your doctor will need to monitor you closely for at least 16 weeks after stopping ULTOMIRIS.
For aHUS, the risks of stopping ULTOMIRIS include an increase in small blood vessel damage, which may cause:
- a large drop in your platelets (thrombocytopenia)
- a large rise in destruction of your red blood cells
- a large drop in your red blood cell count causing anaemia
- decreased urination (problems with your kidneys)
- an increase in your serum creatinine levels (problems with your kidneys)
- confusion or change in how alert you are
- change in your vision
- chest pain or angina
- shortness of breath
- abdominal pain, diarrhoea
- blood clots.
If you experience any of these symptoms, contact your doctor immediately.
- Your doctor will need to monitor you closely.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how ULTOMIRIS affects you.
The effects of this medicine on a person's ability to drive and use machines were not assessed as part of its registration.
Looking after your medicine
ULTOMIRIS will be stored in refrigerated conditions (2°C to 8°C) in the hospital or pharmacy
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What ULTOMIRIS contains
| Active ingredient (main ingredient) | ravulizumab rch (100 mg/mL) |
| Other ingredients (inactive ingredients) |
|
| Potential allergens | Proteins of hamster origin |
Do not take this medicine if you are allergic to any of these ingredients.
What ULTOMIRIS looks like
ULTOMIRIS 100 mg/mL is a translucent, clear to yellowish colour, practically free from particles solution.
(AUST R 330566 - 300 mg/3mL vial)
(AUST R 336710 - 1100 mg/11mL vial).
Who distributes ULTOMIRIS
ULTOMIRIS is registered by:
Alexion Pharmaceuticals Australasia Pty Ltd
Level 4,
66 Talavera Road, Macquarie Park,
NSW 2113
Medical enquiries: 1800 788 189
This leaflet was prepared in August 2025.
Brand Information
| Brand name | Ultomiris 100 mg per mL |
| Active ingredient | Ravulizumab |
| Schedule | S4 |
▼ This medicinal product is subject to additional monitoring in Australia. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse events at www.tga.gov.au/reporting-problems.
MIMS Revision Date: 01 March 2026
Life-threatening meningococcal infections/sepsis have occurred in patients treated with Ultomiris. Meningococcal infection may become rapidly life-threatening or fatal if not recognised and treated early (see Section 4.4 Special Warnings and Precautions for Use).
Immunise patients with meningococcal vaccines at least 2 weeks prior to administering the first dose of Ultomiris, unless the risks of delaying Ultomiris therapy outweigh the risk of developing a meningococcal infection (see Section 4.4 Special Warnings and Precautions for Use for additional guidance on the management of the risk of meningococcal infection).
Refer to the most current edition of the Australian Immunisation Handbook for meningococcal vaccination guidelines.
Patients who initiate Ultomiris treatment less than 2 weeks after receiving meningococcal vaccination must receive treatment with appropriate prophylactic antibiotics until 2 weeks after vaccination.
Vaccination reduces, but does not eliminate, the risk of meningococcal infections. Monitor patients for early signs of meningococcal infections and evaluate immediately if infection is suspected. Patients should be advised about the signs and symptoms of meningococcal infection and to seek medical care immediately if they occur.
1 Name of Medicine
Ravulizumab rch.
2 Qualitative and Quantitative Composition
Ultomiris is a formulation of ravulizumab rch which is a long acting humanised monoclonal immunoglobulin (Ig) G2/4K antibody produced in Chinese hamster ovary (CHO) cell culture by recombinant DNA technology.
Ultomiris is supplied as a single use vial containing 100 mg/mL (300 mg in 3 mL or 1100 mg in 11 mL) of ravulizumab rch.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Concentrated solution for intravenous infusion.
Ultomiris 100 mg/mL is a translucent, clear to yellowish colour, pH 7.4 solution.
4 Clinical Particulars
4.1 Therapeutic Indications
Ultomiris is indicated:
for the treatment of patients with paroxysmal nocturnal haemoglobinuria (PNH);
for the treatment of patients with atypical haemolytic uraemic syndrome (aHUS);
as an add-on to standard therapy for the treatment of adult patients with generalised myasthenia gravis (gMG) who are anti-acetylcholine receptor (AChR) antibody-positive;
for the treatment of adult patients with neuromyelitis optica spectrum disorder (NMOSD) who are anti-aquaporin 4 (AQP4) antibody positive.
Ultomiris is not intended for the acute treatment of a NMOSD relapse.
4.2 Dose and Method of Administration
Dosage. Ultomiris should be administered by a healthcare professional and under appropriate medical supervision.
Adult patients with PNH, aHUS, gMG or NMOSD with body weight ≥ 40 kg. The recommended dosing regimen consists of a loading dose followed by maintenance dosing, administered by intravenous infusion. The doses to be administered are based on the patient's body weight, as shown in Table 1. Maintenance doses should be administered at a once every 8-week interval, starting 2 weeks after loading dose administration.
For patients switching from Soliris (eculizumab rmc) to Ultomiris, the loading dose of Ultomiris should be administered 2 weeks after the last eculizumab rmc maintenance infusion (or 1 week after the last eculizumab rmc induction infusion), and then maintenance doses are administered once every 8 weeks, starting 2 weeks after loading dose administration, as shown in Table 1.
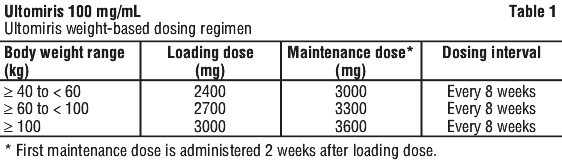
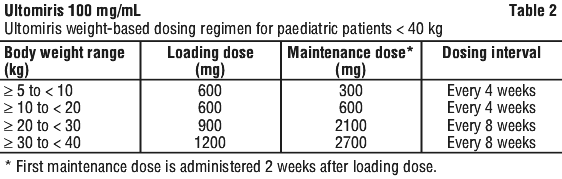
For patients switching from eculizumab to Ultomiris, the loading dose of Ultomiris should be administered 2 weeks after the last eculizumab maintenance infusion (or 1 week after the last eculizumab induction infusion), and then maintenance doses should be administered per weight-based dosing regimen shown in Table 2, starting 2 weeks after loading dose administration.
PNH is a chronic disease and treatment with Ultomiris is recommended to continue for the patient's lifetime, see Section 4.4 Special Warnings and Precautions for Use, Monitoring after Ultomiris discontinuation.
In aHUS, Ultomiris treatment should be a minimum duration of 6 months. Due to the heterogeneous nature of aHUS events and patient-specific risk factors, treatment duration beyond the initial 6 months should be individualised. Patients who are at higher risk for thrombotic microangiopathy (TMA) recurrence, as determined by the treating physician (or clinically indicated) may require chronic therapy, see Section 4.4 Special Warnings and Precautions for Use, Monitoring after Ultomiris discontinuation.
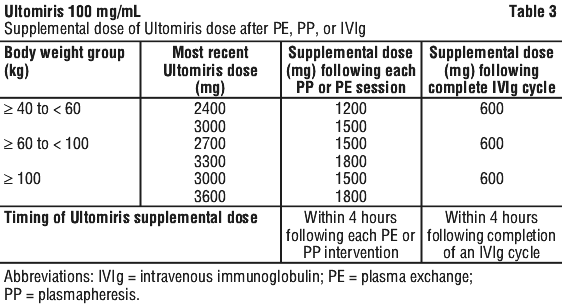
Supplemental dosing following treatment with plasma exchange (PE), plasmapheresis (PP), or intravenous immunoglobulin (IVIg). Plasma exchange (PE), plasmapheresis (PP) and intravenous immunoglobulin (IVIg) have been shown to reduce Ultomiris serum levels. A supplemental dose of Ultomiris is required in the setting of PE, PP or IVIg (see Table 3).
Preparation for administration. Ultomiris must be diluted to a final concentration of 50 mg/mL.
Aseptic technique must be used.
Prepare Ultomiris as follows:
The number of vials to be diluted is determined based on the individual patient's weight and the prescribed dose.
Prior to dilution, the solution in the vials should be visually inspected; the solution should be free of any particulate matter or precipitation. Do not use if there is evidence of particulate matter or precipitation.
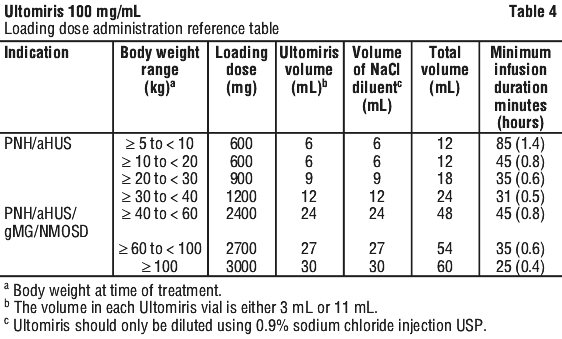
The calculated volume of medicinal product is withdrawn from the appropriate number of vials and diluted in an infusion bag using 0.9% sodium chloride injection USP as diluent. Refer to the administration reference (see Tables 4 and 5).
The product should be mixed gently. It should not be shaken.
After dilution, the final concentration of the solution to be infused is 50 mg/mL.
Each vial of Ultomiris is intended for single use only.
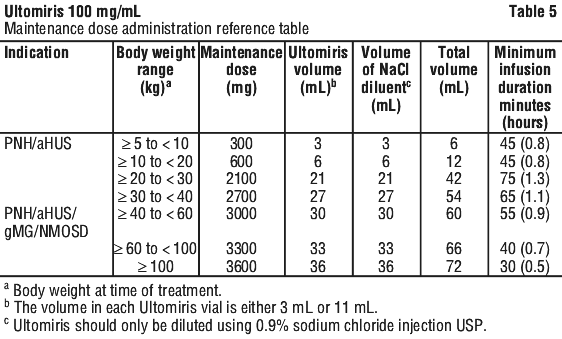
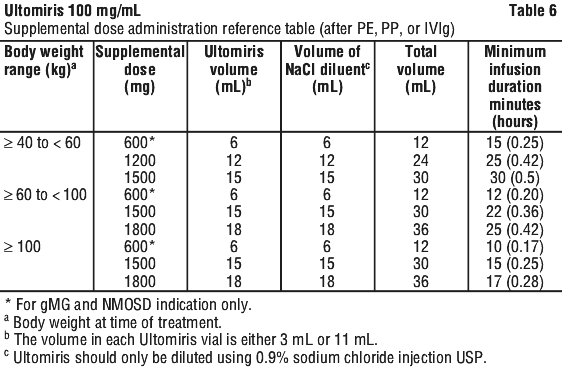
Method of administration. Do not administer as an intravenous push or bolus injection.
The prepared solution should be administered immediately following preparation. Refer to the administration reference tables for minimum infusion duration. If the medicinal product is not used immediately after reconstitution, storage times must not exceed 24 hours at 2°C - 8°C. The prepared solution can be stored for 4 hours at room temperature taking into account the expected infusion time. Infusion must be administered through a 0.2 micrometer filter.
If an adverse reaction occurs during the administration of Ultomiris, the infusion may be slowed or stopped at the discretion of the physician. Patients should be monitored post infusion for signs or symptoms of an infusion-related reaction.
Dosage adjustment. Paediatric population. Ultomiris has not been studied in PNH patients who weigh less than 30 kg. The posology to be used in paediatric patients with PNH who weigh less than 30 kg is identical to the weight-based dosing recommendations provided for paediatric patients with aHUS based on pharmacokinetic/pharmacodynamic (PK/PD) data available in aHUS and PNH patients treated with Ultomiris.
Ultomiris has not been studied in paediatric patients with gMG or NMOSD.
Elderly (> 65 years old). No dose adjustment is required for patients with PNH, aHUS, gMG, or NMOSD aged 65 years and over. There is no evidence indicating any special precautions are required for treating an elderly population although experience with Ultomiris in elderly patients with PNH, aHUS, gMG or NMOSD in clinical studies is limited.
Patients with aplastic anaemia. Ultomiris may be administered to patients with PNH treated with concomitant medications for aplastic anaemia (including immunosuppressive therapies). There is no evidence indicating any special precautions are required for treating patients with aplastic anaemia.
Renal impairment. The clinical trials of Ultomiris in patients with aHUS included patients with renal impairment, some of whom were receiving dialysis. No dose adjustment is required for patients with renal impairment (see Section 5.1 Pharmacodynamic Properties; Section 5.2 Pharmacokinetic Properties, Special populations).
Hepatic impairment. The safety and efficacy of Ultomiris have not been studied in patients with hepatic impairment, however pharmacokinetic data suggest that no dose adjustment is required in patients with hepatic impairment.
4.3 Contraindications
Known hypersensitivity to Ultomiris or to any of the excipients listed in Section 6.1 List of Excipients.
Do not initiate Ultomiris therapy in patients:
with unresolved Neisseria meningitidis infection, see Section 4.4 Special Warnings and Precautions for Use, Serious meningococcal infection;
who are not currently vaccinated against Neisseria meningitidis unless they receive prophylactic treatment with appropriate antibiotics until 2 weeks after vaccination.
4.4 Special Warnings and Precautions for Use
Serious meningococcal infection. Due to its mechanism of action, the use of Ultomiris increases the patient's susceptibility to meningococcal infection/sepsis (Neisseria meningitidis). Meningococcal infection due to any serogroup may occur. To reduce this risk of infection, all patients must be vaccinated against meningococcal infections at least 2 weeks prior to initiating Ultomiris unless the risk of delaying Ultomiris outweighs the risks of developing meningococcal infection. Patients who initiate Ultomiris treatment less than 2 weeks after receiving a meningococcal vaccine must receive treatment with appropriate prophylactic antibiotics until 2 weeks after vaccination. Vaccines against serogroups A, C, Y, W135 and B where available, are recommended to reduce the risk of infection with the commonly pathogenic meningococcal serogroups. Patients must be vaccinated or revaccinated according to current medical guidelines for vaccination use.
Vaccination may not be sufficient to prevent meningococcal infection. Consideration should be given to official guidance on the appropriate use of antibacterial agents. The prescriber and patient should discuss the potential role of ongoing preventative antibacterials. Cases of serious or fatal meningococcal infections/sepsis have been reported in patients treated with Ultomiris and other terminal complement inhibitors. All patients should be monitored for early signs of meningococcal infection and sepsis, evaluated immediately if infection is suspected, and treated with appropriate antibiotics. Patients should be informed of these signs and symptoms and steps should be taken to seek medical care immediately. Physicians should provide patients with a Patient Information Brochure and a Patient Safety Card.
Immunisation. Vaccination may further activate complement. As a result, patients with complement-mediated diseases, may experience increased signs and symptoms of their underlying disease. Therefore, patients should be closely monitored for disease symptoms after recommended vaccination.
Patients below the age of 18 years old must be vaccinated against Haemophilus influenzae and pneumococcal infections and need to adhere strictly to the national vaccination recommendations for their age group.
Other systemic infections. Ultomiris therapy should be administered with caution to patients with active systemic infections. Ultomiris blocks terminal complement activation; therefore, patients may have increased susceptibility to infections, especially infections caused by Neisseria species and encapsulated bacteria. Serious infections with Neisseria species (other than Neisseria meningitidis), including disseminated gonococcal infections, have been reported in patients treated with Ultomiris.
Patients should be provided with a Patient Information Brochure to increase their awareness of potential serious infections and their signs and symptoms.
Physicians should advise patients about gonorrhoea prevention.
Infusion reactions. Administration of Ultomiris may result in systemic infusion reactions and allergic or hypersensitivity reactions (including anaphylaxis). In clinical trials infusion reactions were common (1.9%). They were mild to moderate in severity and transient. These events included back pain, abdominal pain, muscle spasms, drop in blood pressure, elevation in blood pressure, rigors, limb discomfort, drug hypersensitivity (allergic reaction), dysgeusia (bad taste), and drowsiness. In case of systemic infusion reaction, infusion of Ultomiris should be interrupted and appropriate supportive measures should be instituted if signs of cardiovascular instability or respiratory compromise occur (see Section 4.8 Adverse Effects (Undesirable Effects), Post-marketing experience).
Immunogenicity. Treatment with any therapeutic protein may induce an immune response. In Ultomiris studies (PNH studies [n = 488], aHUS studies [n = 92], a gMG study [n = 167] and a NMOSD study [n = 58]), treatment-emergent anti-drug antibodies were reported in 10 patients (1.2%), 6 with PNH, 2 with aHUS and 2 with gMG. Of these, only 2 (PNH) were persistent, at low titres, and neither correlated with clinical response or adverse events.
Monitoring after Ultomiris discontinuation. Treatment discontinuation for PNH. PNH is a chronic disease and treatment with Ultomiris is recommended to continue for the patient's lifetime.
If patients with PNH discontinue treatment with Ultomiris, they should be closely monitored for signs and symptoms of haemolysis, identified by elevated lactate dehydrogenase (LDH) along with sudden decrease in PNH clone size or haemoglobin, or re-appearance of symptoms such as fatigue, haemoglobinuria, abdominal pain, shortness of breath (dyspnoea), major adverse vascular event (including thrombosis), dysphagia, or erectile dysfunction. Any patient who discontinues Ultomiris should be monitored for at least 16 weeks to detect haemolysis and other reactions. If signs and symptoms of haemolysis occur after discontinuation, including elevated LDH, consider restarting treatment with Ultomiris.
Treatment discontinuation for aHUS. There are no specific data on Ultomiris discontinuation. Severe TMA complications were observed following Soliris discontinuation in aHUS clinical studies (in some patients up to 127 weeks after discontinuation) and can occur at any time.
If patients must discontinue treatment with Ultomiris, they should be closely monitored for signs and symptoms of TMA on an on-going basis. Monitoring may be insufficient to predict or prevent TMA complications.
TMA complications post-discontinuation can be identified if any of the following is observed:
(i) At least two of the following laboratory results observed concurrently: a decrease in platelet count of 25% or more as compared to either baseline or to peak platelet count during Ultomiris treatment; an increase in serum creatinine of 25% or more as compared to baseline or to nadir during Ultomiris treatment; or, an increase in serum LDH of 25% or more as compared to baseline or to nadir during Ultomiris treatment (results should be confirmed by a second measurement).
(ii) Any one of the following symptoms of TMA: a change in mental status or seizures or other extra-renal TMA manifestations including cardiovascular abnormalities, pericarditis, gastrointestinal symptoms/diarrhoea; or thrombosis.
If TMA complications occur after Ultomiris discontinuation, re-initiation of Ultomiris treatment should be considered, beginning with the loading dose and maintenance dose (see Section 4.2 Dose and Method of Administration).
Treatment discontinuation for gMG. As gMG is a chronic disease, patients benefiting from Ultomiris who discontinue treatment should be monitored for symptoms of the underlying disease. If symptoms of gMG occur after discontinuation, consider restarting treatment with Ultomiris.
Treatment discontinuation for NMOSD. As NMOSD is a chronic disease, patients benefiting from Ultomiris treatment who discontinue treatment should be monitored for symptoms of NMOSD relapse. If symptoms of NMOSD relapse occur after discontinuation, consider restarting treatment with Ultomiris.
Use in the elderly. Ultomiris may be administered to patients with PNH, aHUS, gMG or NMOSD aged 65 years and over. There is no evidence indicating any special precautions are required for treating an elderly population.
Paediatric use. Based on data available in aHUS and PNH patients treated with Ultomiris, the efficacy and safety profile in paediatric patients with body weight ≥ 5 kg is expected to be similar to that of adults.
Use of Ultomiris in paediatric PNH patients with body weight < 30 kg is based on extrapolation of PK/PD, efficacy and safety data from aHUS and PNH clinical studies.
Ultomiris has not been studied in paediatric patients with gMG or NMOSD.
Effects on laboratory tests. No data available.
4.5 Interactions with Other Medicines and Other Forms of Interactions
No interaction studies have been performed.
Concomitant use of Ultomiris with neonatal Fc receptor (FcRn) blockers may lower systemic exposures and reduce effectiveness of Ultomiris. Closely monitor for reduced effectiveness of Ultomiris. See Section 4.2 for guidance in case of concomitant PE, PP, or IVIg treatment.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. No studies on fertility have been conducted specifically with Ultomiris.
A study in mice with a surrogate terminal complement inhibitor (murine anti-C5) antibody identified no adverse effect on fertility of the treated females or males. Use of a surrogate molecule was required as Ultomiris does not recognise the form of the pharmacological target present in laboratory animal species.
Use in pregnancy. (Category B2)
No clinical data on exposed pregnancies are available.
No studies on embryofetal development have been conducted specifically with Ultomiris. A study in mice with a murine surrogate terminal complement inhibitory (anti-C5) antibody given during the period of organogenesis identified no clear treatment-related findings in fetuses of mice exposed to 60 mg/kg/week but is of limited predictive value. When exposure to the murine antibody occurred from the time of implantation to the end of lactation, a slightly higher number of male offspring became moribund or died in the group given 60 mg/kg/week. The relevance to use of Ultomiris is unclear. Human IgG are known to cross the human placental barrier, and thus Ultomiris may potentially cause terminal complement inhibition in the fetal circulation.
Women of childbearing potential should use effective contraception methods during treatment and for 8 months after treatment.
Use in lactation. It is unknown whether Ultomiris is excreted into human milk. Since many medicinal products and immunoglobulins are secreted into human milk, and because of the potential for serious adverse reactions in nursing infants, breast-feeding should be discontinued during treatment and for 8 months after treatment.
4.7 Effects on Ability to Drive and Use Machines
No studies on the effects on the ability to drive and use machines have been performed.
4.8 Adverse Effects (Undesirable Effects)
Clinical trial experience. The most common adverse drug reactions across all clinical trials are headache, upper respiratory tract infection, nasopharyngitis, diarrhoea, pyrexia, nausea, arthralgia, back pain, fatigue, abdominal pain, urinary tract infection and dizziness. The most serious adverse reactions in patients in clinical trials were meningococcal infection and meningococcal sepsis.
The clinical safety data described below were obtained with a lower concentration (10 mg/mL) formulation of Ultomiris.
Table 7 gives the adverse reactions observed from clinical trials.
Adverse reactions are listed by MedDRA system organ class (SOC) and frequency, using the following convention: very common (≥ 1/ 10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000); and not known (cannot be estimated from available data).
Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness.
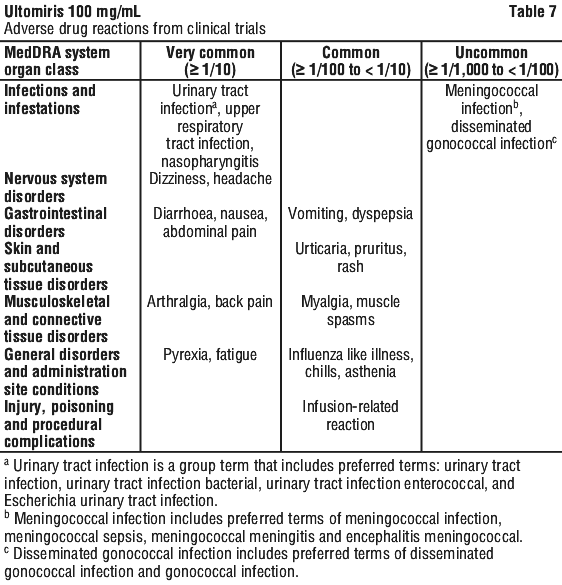
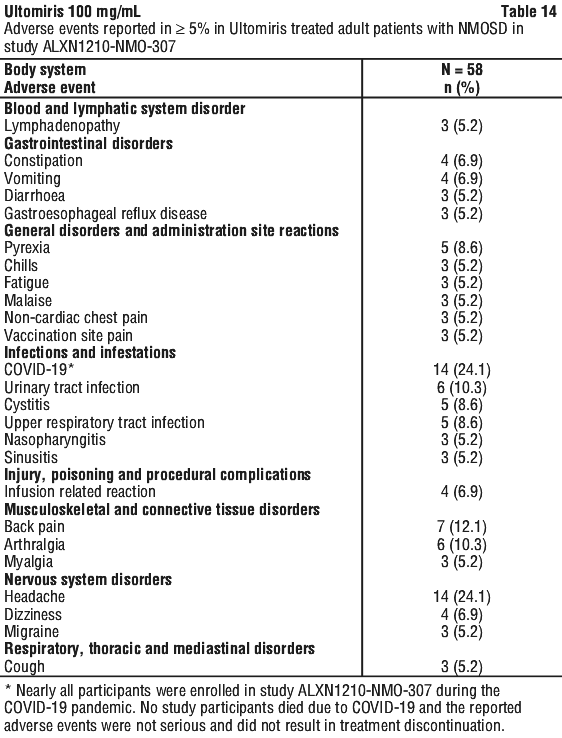
Table 8 describes adverse events that occurred at a rate of 5% or more among patients treated with Ultomiris in PNH studies.
Serious adverse events were reported in 15 (6.8%) patients with PNH receiving Ultomiris. The serious adverse events in patients treated with Ultomiris included hyperthermia and pyrexia. No serious adverse events were reported in more than 1 patient treated with Ultomiris.
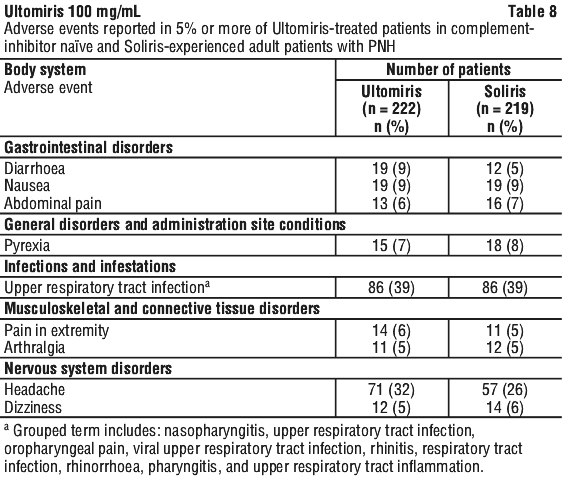
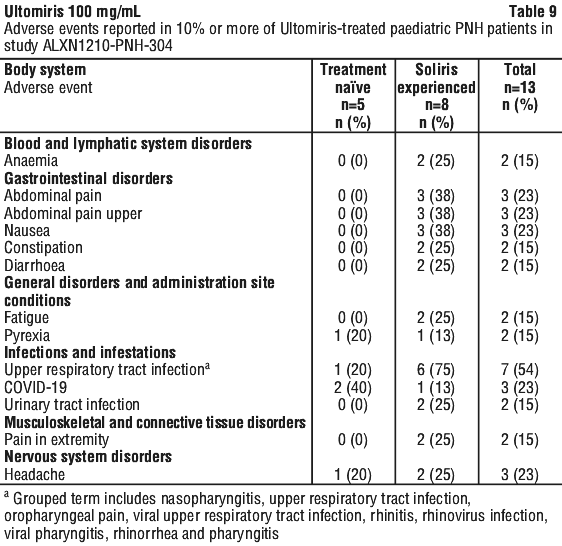
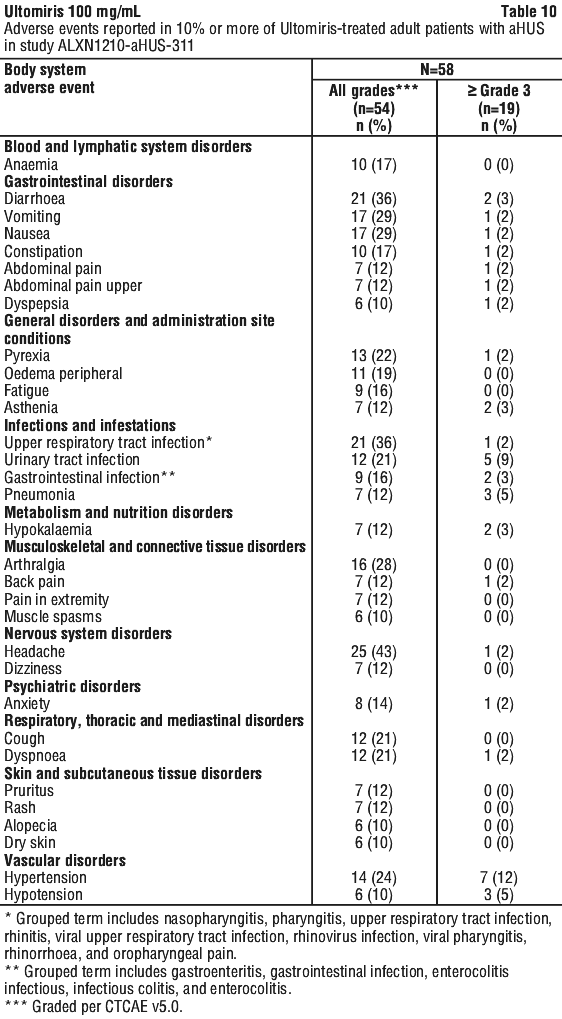
Table 10, Table 11 and Table 12 describe the adverse events that occurred at a rate of 10% or more among patients treated with Ultomiris in the aHUS studies.
Serious adverse events were reported in 54 (65.85%) patients with aHUS receiving Ultomiris. The most frequent serious adverse events reported in more than 3 patients (3.3%) treated with Ultomiris were hypertension, pneumonia, diarrhoea and pyrexia. Five patients died during the ALXN1210-aHUS-311 study. The cause of death was sepsis in 2 patients, intracranial haemorrhage in 1 patient, pre-treatment cerebral arterial thrombosis in 1 patient that was excluded from the trial after a diagnosis of Shiga toxin Escherichia coli related haemolytic uraemic syndrome (STEC-HUS) and acute cardiac failure in 1 patient during the extension period.
Generalised myasthenia gravis (gMG). The safety of Ultomiris has been evaluated in 175 adult patients with gMG, including 169 patients who received at least one dose of Ultomiris, 160 patients who were exposed for at least 6 months, and 150 who were exposed for at least 12 months (see Section 5.1 Pharmacodynamic Properties, Clinical trials, Generalised myasthenia gravis (gMG)). In a randomised, double-blind, placebo-controlled trial (ALXN1210-MG-306), the most frequent adverse events (> 10%) with Ultomiris were diarrhoea and upper respiratory tract infection. Table 13 describes adverse events that occurred at a rate of 5% or more and at greater frequency than placebo.
Serious adverse events were reported in 20 (23%) patients with gMG receiving Ultomiris and in 14 (16%) patients receiving placebo. The most frequent serious adverse events were infections reported in at least 8 (9%) patients treated with Ultomiris and in 4 (4%) patients treated with placebo. Of these infections, one fatal case of COVID-19 pneumonia was identified in a patient treated with Ultomiris and one case of infection led to the discontinuation of Ultomiris.

Post-marketing experience. Infusion reactions. Administration of Ultomiris may result in infusion reactions and allergic or hypersensitivity reactions (including anaphylaxis). In case of infusion reaction, infusion of Ultomiris should be interrupted and appropriate supportive measures should be instituted if signs of cardiovascular instability or respiratory compromise occur.
Urticaria. Urticaria has been noted as a common adverse event in patients treated with Ultomiris.
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
4.9 Overdose
No case of overdose has been reported to date.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
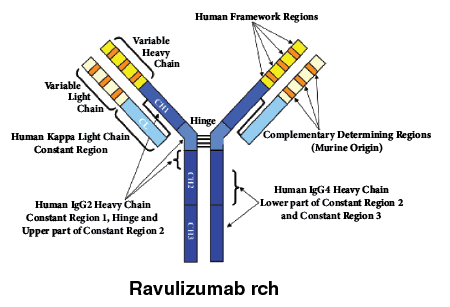
Ravulizumab rch is a humanised monoclonal antibody (mAb) consisting of 2 identical 448 amino acid heavy chains and 2 identical 214 amino acid light chains and has a molecular weight of approximately 148 kDa. The constant regions of ravulizumab rch include the human kappa light chain constant region, and the protein engineered "IgG2/4" heavy chain constant region.
The heavy chain CH1 domain, hinge region, and the first 5 amino acids of the CH2 domain match the human IgG2 amino acid sequence, residues 6 to 36 in the CH2 region (common to both human IgG2 and IgG4 amino acid sequences), while the remainder of the CH2 domain and the CH3 domain match the human IgG4 amino acid sequence. The heavy and light chain variable regions that form the human C5 binding site consist of human framework regions grafted to murine complementarity-determining regions.
Mechanism of action. Ravulizumab rch is a terminal complement inhibitor that specifically binds to the complement protein C5 with high affinity, thereby inhibiting its cleavage to C5a (the pro-inflammatory anaphylatoxin) and C5b (the initiating subunit of the terminal complement complex [C5b-9, also known as the membrane attack complex (MAC)]), thus preventing MAC formation. By binding specifically to C5, ravulizumab rch antagonises terminal complement-mediated inflammation, cell activation, and cell lysis while preserving the early components of complement activation that are essential for opsonisation of microorganisms and clearance of immune complexes.
This mechanism of action provides the therapeutic rationale for the use of ravulizumab rch in PNH, aHUS, gMG and NMOSD, in which uncontrolled complement activation is involved. In patients with PNH, complement-mediated intravascular haemolysis is blocked with ravulizumab rch treatment. Ravulizumab rch also resolves the complement-mediated TMA present in aHUS. In patients with gMG, ravulizumab rch inhibits terminal complement activation which otherwise leads to MAC deposition at the neuromuscular junction resulting in impairment of neuromuscular transmission. In NMOSD, ravulizumab rch inhibits C5a dependant inflammation and MAC formation that otherwise leads to astrocytopathy and damage to surrounding glial cells and neurons.
Ravulizumab rch was specifically engineered to dissociate from C5 and associate with human neonatal Fc receptor (FcRn) at pH 6.0 (while minimising the impact in binding to C5 in intravascular space where the normal pH is 7.4). As a result, dissociation of antibody:C5 complexes in the acidified environment of the early endosome after pinocytosis is increased. Therefore, free antibody is recycled from the early endosome back into the vascular compartment by FcRn, resulting in an extended ravulizumab rch terminal elimination half-life (see Section 5.2 Pharmacokinetic Properties).
Ravulizumab rch dosing has been optimised to achieve therapeutic steady state concentrations following the first dose, resulting in immediate onset of action and complete terminal complement inhibition by the end of infusion; ravulizumab rch half-life in serum yields prolonged pharmacologic activity, allowing dosing once every 8 weeks (or once every 4 weeks for patients weighing less than 20 kg).
Pharmacodynamic effects. Following ravulizumab rch treatment in both adult and paediatric complement-inhibitor naïve patients and eculizumab rmc-experienced patients with PNH in Phase 3 studies, immediate and complete inhibition of serum free C5 (concentration of < 0.5 microgram/mL) was observed by the end of the first infusion and sustained throughout the entire 26-week treatment period (Figure 1 and Figure 2). In contrast, serum free C5 concentrations did not consistently remain < 0.5 microgram/mL following eculizumab rmc treatment (Figure 1 and Figure 2).
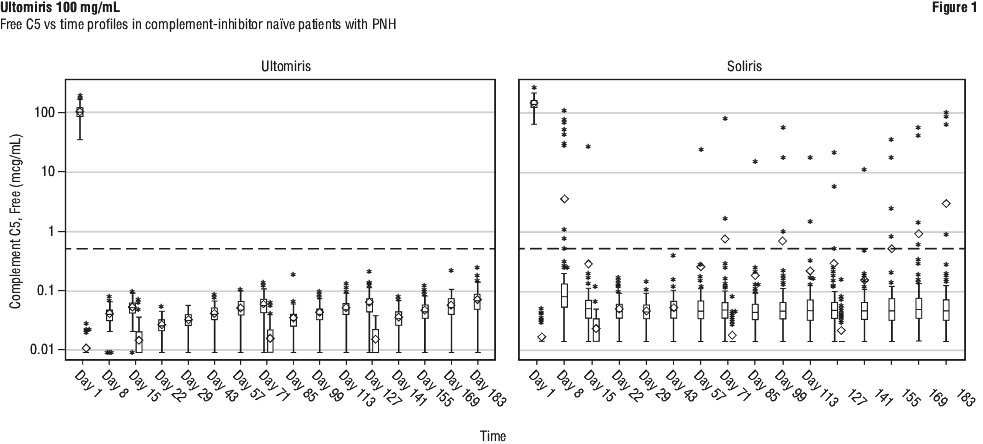
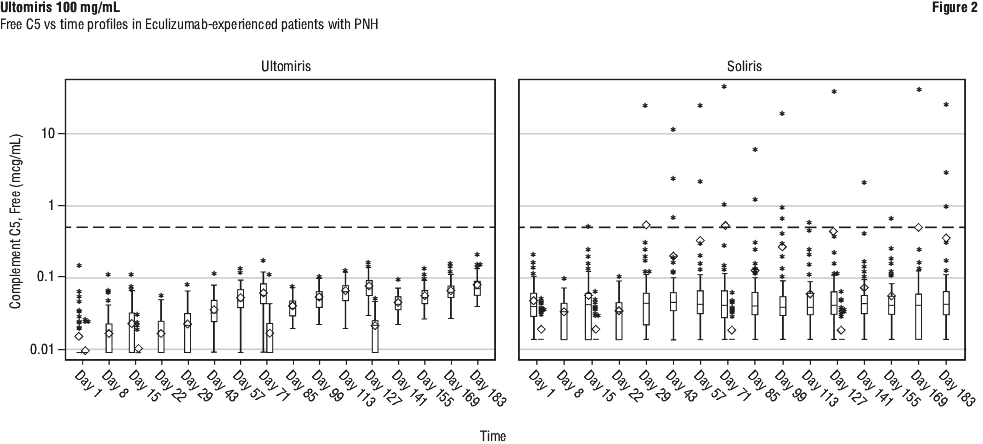
Following ravulizumab rch treatment administration, immediate and complete inhibition of serum free C5 was also observed in adult and paediatric patients with aHUS and adult patients with gMG and NMOSD by the end of the first infusion and was sustained throughout the primary treatment period.
The extent and duration of the pharmacodynamic response were exposure-dependent in patients with PNH, aHUS, gMG and NMOSD following ravulizumab rch treatment.
Clinical trials. Paroxysmal nocturnal haemoglobinuria (PNH). The clinical development program was designed to determine whether Ultomiris is non-inferior to the current standard of care therapy, Soliris (eculizumab rmc) in adult patients with PNH regardless of previous treatment status while assessing potential beneficial effects of a longer dosing interval. The safety and efficacy of Ultomiris in patients with PNH were assessed in 2 distinct and complementary populations: a complement-inhibitor-naive population of patients with active haemolysis to establish the magnitude of the efficacy response, and a population of patients stable on Soliris therapy that allowed the assessment of the maintenance of efficacy and safety in a population switching to Ultomiris.
Accordingly, 2 adequate and well controlled Phase 3 trials were conducted to cover each population:
a Complement-Inhibitor Naive Study in adult patients with PNH who were naive to complement inhibitor treatment (ALXN1210-PNH-301);
a Soliris-Experienced Study in patients with PNH who were clinically stable after having been treated with Soliris (eculizumab rmc) for at least the previous 6 months (ALXN1210-PNH-302).
Ultomiris was dosed in accordance with the recommended dosing described in Section 4.2 Dose and Method of Administration (4 infusions of Ultomiris over 26 weeks) while Soliris was administered according to the approved dosing regimen of Soliris (15 infusions over 26 weeks) which was the standard-of-care for PNH at the time of studies.
Patients were vaccinated against meningococcal infection prior to, or at the time of initiating treatment with Ultomiris or Soliris, or received prophylactic treatment with appropriate antibiotics until 2 weeks after vaccination.
There were no noteworthy differences in the demographic or baseline characteristics between the Ultomiris and Soliris treatment groups in either of the Phase 3 studies. Twelve-month transfusion history was similar between Ultomiris and Soliris treatment groups within each of the Phase 3 studies.
ALXN1210-PNH-301 study in complement-inhibitor naive adult patients with PNH. The Complement-Inhibitor Naive Study was a 26-week, multicentre, open-label, randomised, active-controlled, Phase 3 study conducted in 246 patients who were naive to complement inhibitor treatment prior to study entry and was followed by a long-term extension period where all patients received Ultomiris.
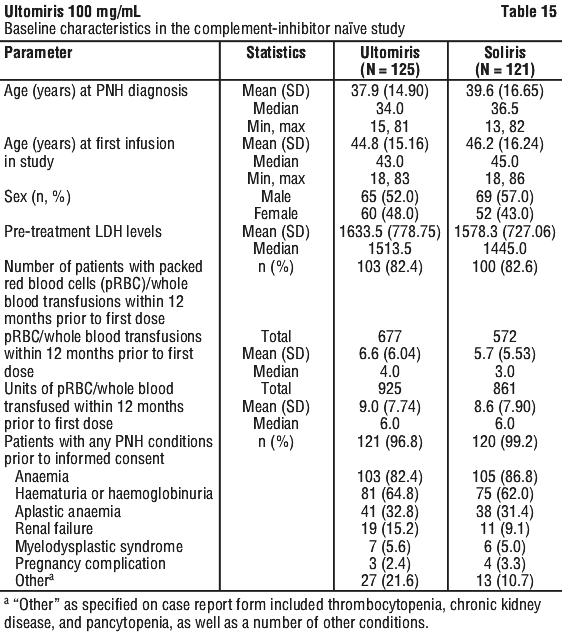
PNH medical history was similar between Ultomiris and Soliris treatment groups. The 12-month transfusion history was similar between Ultomiris and Soliris treatment groups. More than 80% of patients in both treatment groups had a history of transfusion within 12 months of study entry. The majority of the Complement-Inhibitor Naive Study population was highly haemolytic at baseline; 86.2% of enrolled patients presented with elevated LDH ≥ 3 x ULN, which is a direct measurement of intravascular haemolysis, in the setting of PNH. The median total red blood cell (RBC) clone size was 33.75%, consistent with ongoing active haemolysis of PNH erythrocytes in a patient population with a large median granulocyte clone size (92.55%).
Table 15 presents the baseline characteristics of the patients with PNH enrolled in the Complement-Inhibitor Naive Study.
In the Complement-Inhibitor Naive Study, both co-primary endpoints, avoidance of packed red blood cells (pRBC) transfusion per protocol-specified guidelines, and LDH normalisation from Day 29 to Day 183, met the primary objective and showed Ultomiris was statistically significant for non-inferiority compared to Soliris. Ultomiris also achieved statistically significant non-inferiority compared to Soliris for all 4 key secondary endpoints. Both co-primary endpoints and all key secondary endpoints favoured Ultomiris (Figure 3).

The mean percent change in LDH from baseline to Day 183 was -76.84% for the Ultomiris group and -76.02% for the Soliris group. The mean difference between treatment groups was -0.83% (95% CI: -5.21%, 3.56%).
The mean change in FACIT-Fatigue total score from baseline to Day 183 was 7.07 for the Ultomiris group and 6.40 for the Soliris group, with a 3-point improvement from baseline on this scale considered a clinically meaningful improvement. The mean difference between treatment groups was 0.67 (95% CI: -1.21, 2.55). Both treatment groups showed improvement in fatigue as measured by FACIT-Fatigue overtime. Improvement was numerically greater with Ultomiris than Soliris at all time points for FACIT-Fatigue.
Breakthrough haemolysis defined as at least 1 new or worsening symptom or sign of intravascular haemolysis in the presence of elevated LDH ≥ 2 x ULN, after prior LDH reduction to < 1.5 x ULN on therapy, was experienced by 4.0% of patients in the Ultomiris group and 10.7% of patients in the Soliris group. The difference between treatment groups was -6.7% (95% CI: -14.21%, 0.18%).
Haemoglobin stabilisation defined as an avoidance of a ≥ 2 g/dL decrease in haemoglobin level from baseline in the absence of transfusion through Day 183 was achieved by 68.0% of patients in the Ultomiris group and 64.5% of patients in the Soliris group. The difference between treatment groups was 2.9% (95% CI: -8.80%, 14.64%).
Because statistically significant non-inferiority was achieved for both co-primary and all 4 key secondary endpoints, superiority was assessed following the pre-specified hierarchical testing order that began with the breakthrough haemolysis endpoint. The treatment difference for breakthrough haemolysis (p = 0.0558) did not reach the pre-specified threshold for superiority (p < 0.05), and no further testing was conducted. The incidence of breakthrough haemolysis was more than 2-fold higher in the Soliris group (13 patients with 15 events) than in the Ultomiris group (5 patients with 5 events). Of the 15 breakthrough haemolysis events seen in the Soliris group, 7 were associated with elevated free C5 above 0.5 microgram/mL. No patients in the Ultomiris group had elevations of free C5 levels above 0.5 microgram/mL.
The final efficacy analysis for the study included all patients ever treated with Ultomiris (n=244) and had a median treatment duration of 203.3 weeks (1423 days). The final analysis confirmed that Ultomiris treatment responses observed during the primary evaluation period (through week 26) were maintained throughout the duration of the study.
ALXN1210-PNH-302 study in adult PNH patients previously treated with Soliris. The Soliris-Experienced Study was a 26-week, multicentre, open-label, randomised, active-controlled Phase 3 study conducted in 195 patients with PNH who were clinically stable after having been treated with Soliris (eculizumab rmc) for at least the past 6 months and was followed by a long-term extension period where all patients received Ultomiris.
PNH medical history was similar between Ultomiris and Soliris treatment groups. The 12-month transfusion history was similar between Ultomiris and Soliris treatment groups and more than 87% of patients in both treatment groups had not received a transfusion within 12 months of study entry. Per study entry criteria, all patients presented with controlled haemolysis at baseline, consistent with a population under continuous treatment with Soliris. The mean total PNH RBC clone size was 60.05%, the mean total PNH granulocyte clone size was 83.30%, and the mean total PNH monocyte clone size was 85.86%.
Table 16 presents the baseline characteristics of the PNH patients enrolled in the Soliris-Experienced Study.
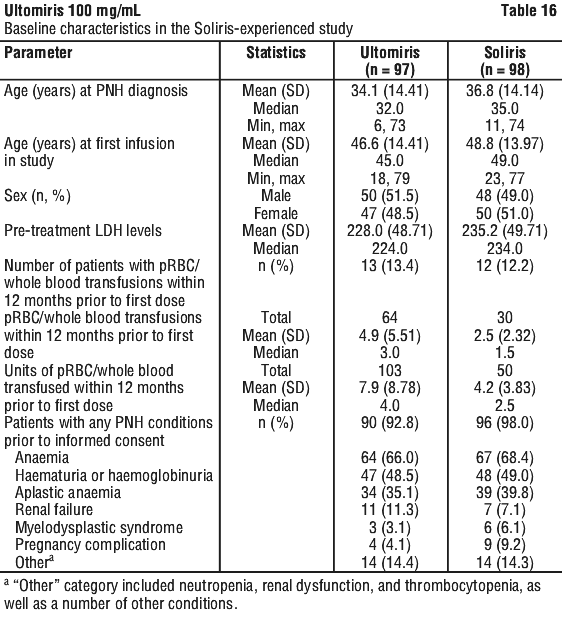
In the Soliris-Experienced Study, the primary endpoint, percent change in LDH from baseline to Day 183, met the primary objective and showed Ultomiris was statistically significant for non-inferiority compared to Soliris. Ultomiris also achieved statistically significant non-inferiority compared to Soliris for all 4 key secondary endpoints. Both the primary endpoints and all key secondary endpoints favoured Ultomiris (Figure 4).
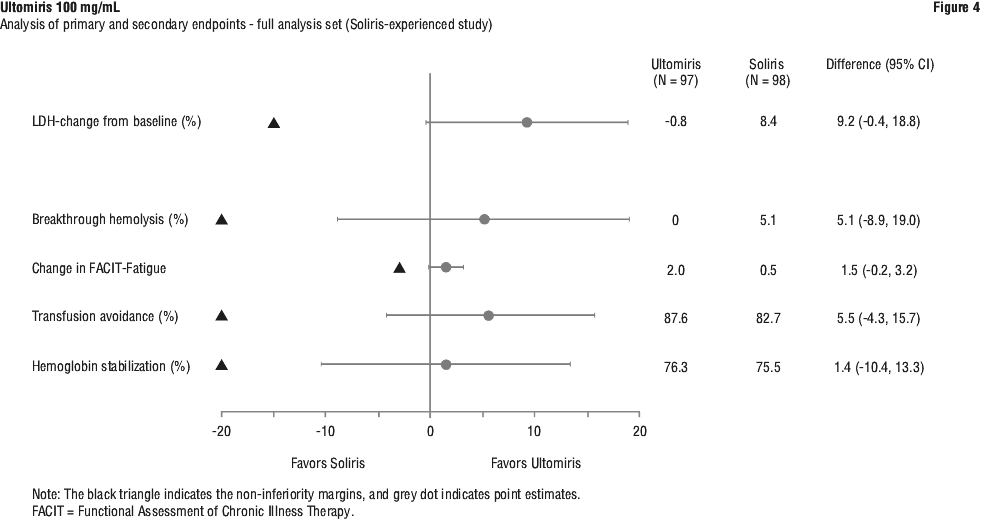
Breakthrough haemolysis, using the same definition as the Complement-Inhibitor Naive Study, was experienced by none of the patients in the Ultomiris group and 5 (5.1%) of the patients in the Soliris group. The difference between treatment groups was -5.1% (95% CI: -18.99%, 8.89%). The incidence of breakthrough haemolysis was higher in the Soliris group (7 events) than in the Ultomiris group (0 events). Of the 7 breakthrough haemolysis events seen in the Soliris group, 4 were associated with elevated free C5 above 0.5 microgram/mL. There were no breakthrough haemolysis events in the Ultomiris group and no patients in the Ultomiris group had elevations of free C5 levels above 0.5 microgram/mL.
The mean change in the FACIT-Fatigue total score from baseline to Day 183 was 2.01 for the Ultomiris group and 0.54 for the Soliris group. The LS mean difference between treatment groups was 1.5 (95% CI: -0.2, 3.2). Both treatment groups showed improvement in fatigue as measured by FACIT-Fatigue over time; improvement was numerically greater with Ultomiris than Soliris at all time points for the FACIT-Fatigue following Day 8.
Transfusion avoidance was achieved by 87.6% of patients on Ultomiris compared to 82.7% of patients on Soliris by Week 26. The difference between the Ultomiris and Soliris treatment groups in the percentage of patients who avoided transfusion was 5.5% (95% CI: -4.27%, 15.68%).
Haemoglobin stabilisation through Day 183 was achieved by 76.3% of patients in the Ultomiris group and 75.5% of patients in the Soliris group. The difference between treatment groups was 1.4% (95% CI: -10.41%, 13.31%).
As statistically significant non-inferiority was achieved for the primary endpoint and all 4 key secondary endpoints, the pre-specified hierarchical order continued with superiority testing of percent change from baseline in LDH. The assessment of the treatment difference for superiority resulted in a p-value of 0.0583 which did not reach the pre-specified significance threshold for superiority (p < 0.05) and therefore no additional testing in the hierarchy was conducted.
Overall, treatment with Ultomiris in both complement-inhibitor naive and Soliris-experienced patients was associated with clinically meaningful benefits across disease-relevant endpoints and reduction of the overall risk of breakthrough haemolysis through better C5 control and elimination of the risk of pharmacodynamic-associated breakthrough haemolysis.
The final efficacy analysis for the study included all patients ever treated with Ultomiris (n=192) and had a median treatment duration of 138.3 weeks (968 days). The final analysis confirmed that Ultomiris treatment responses observed during the primary evaluation period (through week 26) were maintained throughout the duration of the study.
ALXN1210-PNH-304 study in paediatric patients with PNH. The paediatric study was a multicentre, open-label, Phase 3 study conducted in Soliris-experienced and complement inhibitor treatment naive paediatric patients with PNH. Patients who completed the 26-week primary evaluation period were followed for up to 4 years in the long-term extension period.
A total of 13 PNH paediatric patients completed Ultomiris treatment during the Primary Evaluation Period (26 weeks) of Study ALXN1210-PNH-304. Five of the 13 patients had never been treated with complement inhibitors and 8 patients were treated with Soliris (eculizumab rmc). Eleven of the 13 patients were between 12 and 17 years of age at first infusion, with 2 patients under 12 years old (11 and 9 years old). Based on body weight, patients received a loading dose of Ultomiris on Day 1, followed by maintenance treatment on Day 15 and once every 8 weeks (q8w) thereafter for patients weighing ≥ 20 kg, or once every 4 weeks (q4w) for patients weighing < 20 kg. For patients who entered the study on Soliris therapy, Day 1 of study treatment was planned to occur 2 weeks from the patient's last dose of Soliris.
Table 17 presents the baseline characteristics of the PNH patients enrolled in the Paediatric PNH Study.
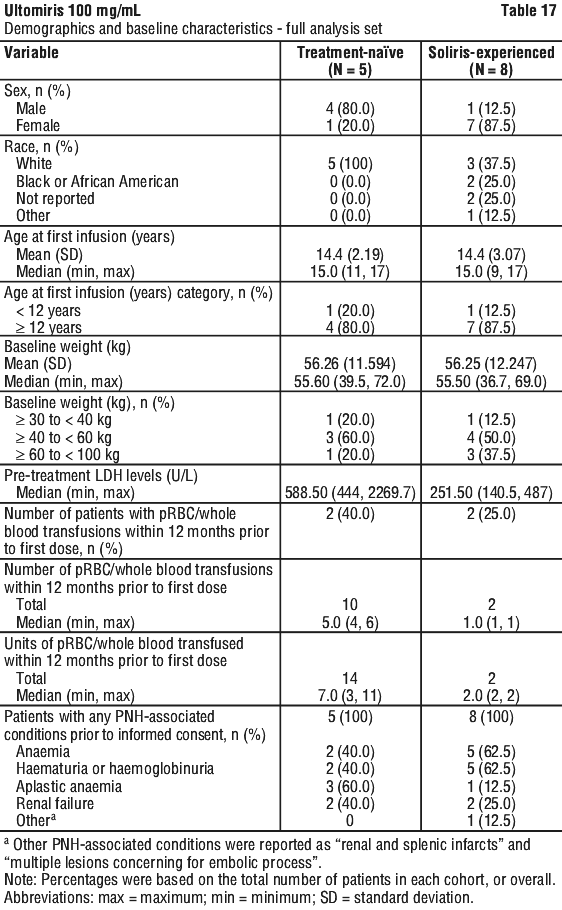
Table 18 presents secondary efficacy outcomes for the Primary Evaluation Period.
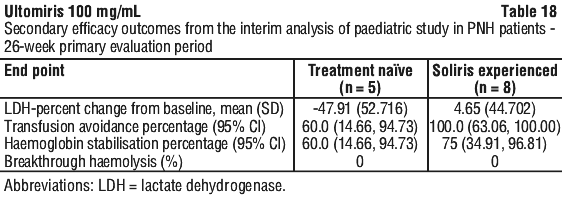
The efficacy of Ultomiris in paediatric PNH patients appears to be similar to that observed in adult PNH patients enrolled in pivotal studies.
Atypical haemolytic uraemic syndrome (aHUS). The safety and efficacy of Ultomiris in patients with aHUS were assessed in 2 open-label, single arm, Phase 3 studies. Study ALXN1210-aHUS-311 enrolled adult patients with aHUS and Study ALXN1210-aHUS-312 enrolled paediatric patients with aHUS.
ALXN1210-aHUS-311 study in adult patients with aHUS. The adult study was a multicentre, single arm, Phase 3 study conducted in patients who were naive to complement inhibitor treatment prior to study entry and had evidence of TMA. The study consisted of a 26-week Initial Evaluation Period and patients were allowed to enter an extension period for up to 4.5 years.
A total of 58 patients with documented aHUS were enrolled. Enrolment criteria excluded patients presenting with TMA due to a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) deficiency, STEC-HUS and genetic defect in cobalamin C metabolism. Two patients were excluded from the Full Analysis Set due to a confirmed diagnosis of STEC-HUS. The majority of patients (92.9%) had extra renal signs or symptoms of aHUS at baseline. At baseline, 72.2% (n = 39) of patients had Stage 5 chronic kidney disease (CKD).
Table 19 presents the demographics and baseline characteristics of the 56 adult patients enrolled in Study ALXN1210-aHUS-311 that constituted the Full Analysis Set.
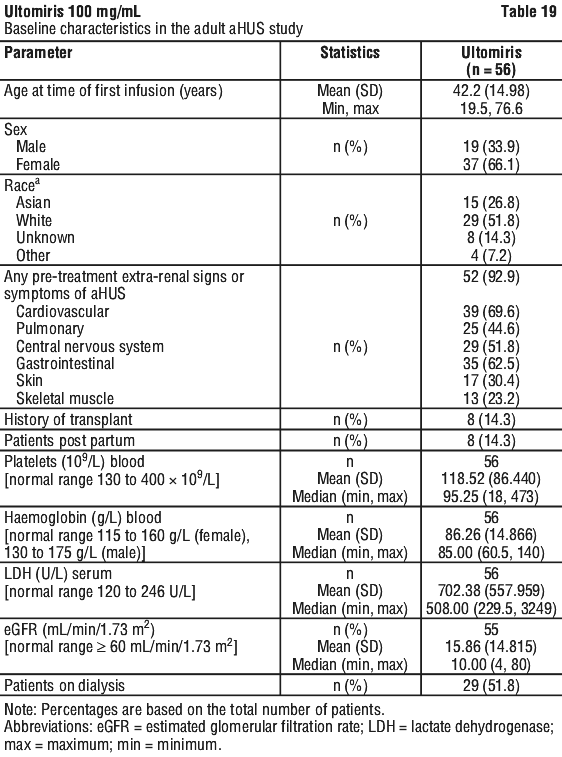
Complete TMA Response was observed in 30 of the 56 patients (53.6%) during the 26-week Initial Evaluation Period as shown in Table 20.
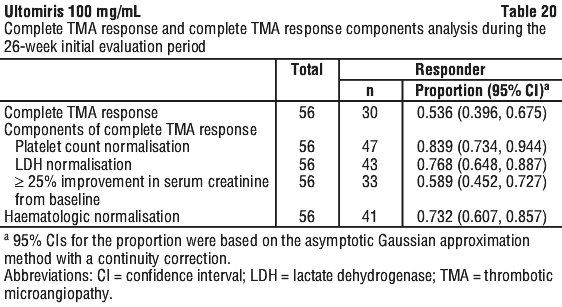
The median time to Complete TMA Response was 86 days (7 to 1359 days). A rapid increase in mean platelet count was observed after commencement of Ultomiris, increasing from 118.52 x 109/L at baseline to 243.54 x 109/L at Day 8 and remaining above 227 x 109/L at all subsequent visits in the Initial Evaluation Period (26 weeks). Similarly, the mean LDH value decreased from baseline over the first 2 months of treatment and was sustained over the duration of the Initial Evaluation Period (26 weeks). Table 21 summarises the secondary efficacy results for Study ALXN1210-aHUS-311.
Renal function, as measured by eGFR, was improved or maintained during Ultomiris treatment. By Day 743 of the study, over two thirds of the remaining patient population (26/36), who were mostly CKD Stage 4 or 5 at baseline, improved their CKD stages. Chronic kidney disease stage continued to improve for many patients (19/30) after achieving Complete TMA Response during the 26-week Initial Evaluation Period. Eighteen of the 25 patients who required dialysis at study entry were able to discontinue dialysis by the end of the Initial Evaluation Period, with 2 additional patients discontinuing dialysis during the Extension Period. While 8 participants who were off dialysis at baseline-initiated dialysis during the study.

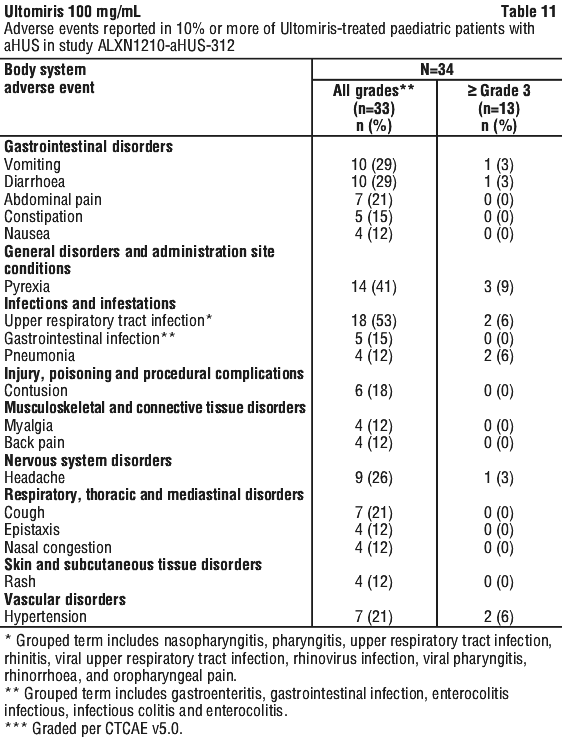
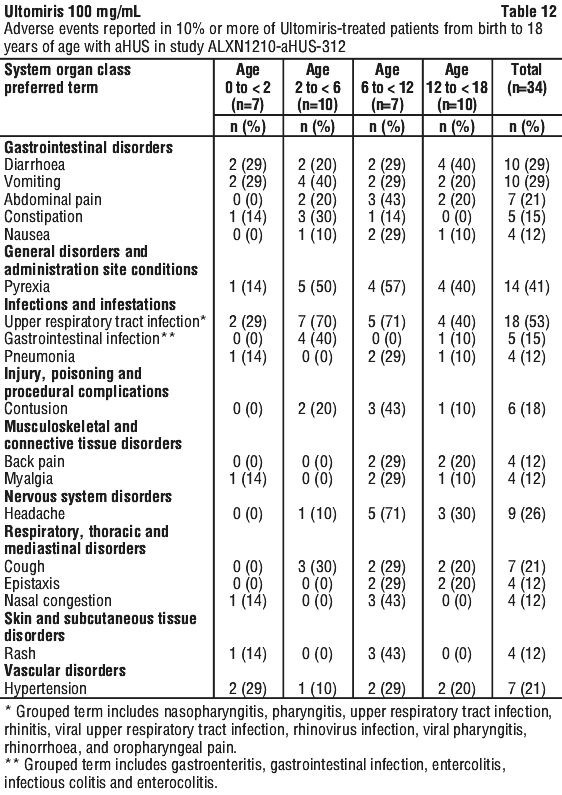
ALXN1210-aHUS-312 study in paediatric patients with aHUS. Study ALXN1210-aHUS-312 was a 26-week, multicentre, single arm, Phase 3 study conducted in paediatric patients. After the 26-week Initial Evaluation Period, patients were allowed to enter an Extension Period for up to 4.5 years.
A total of 24 Soliris-naive patients with documented diagnosis of aHUS and evidence of TMA were enrolled, of whom 20 were included in the Full Analysis Set. The median age at the time of first infusion was 4.8 years (range: 0.9, 17.3 years). Enrolment criteria excluded patients presenting with TMA due to a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) deficiency, STEC-HUS and genetic defect in cobalamin C metabolism. Four patients were given 1 or 2 doses but were excluded from the study because eligibility criteria were not confirmed for aHUS diagnosis.
The overall mean weight at baseline was 21.2 kg; the majority of patients (55%) were in the baseline weight category ≥ 10 to < 20 kg. The majority of patients (70.0%) had pre-treatment extra renal signs (cardiovascular, pulmonary, central nervous system, gastrointestinal, skin, skeletal muscle) or symptoms of aHUS at baseline. At baseline, 35.0% (n = 7) of patients had CKD Stage 5.
A total of 10 patients who switched from Soliris to Ultomiris with documented diagnosis of aHUS were enrolled. Patients had to have a clinical response to Soliris prior to enrolment i.e. LDH < 1.5 x ULN and platelet count ≥ 150,000/microliter, and eGFR > 30 mL/min/1.73 m2). Consequently, there is no information on the use of Ultomiris in patients refractory to Soliris.
Table 22 presents the baseline characteristics of the paediatric patients enrolled in Study ALXN1210-aHUS-312.
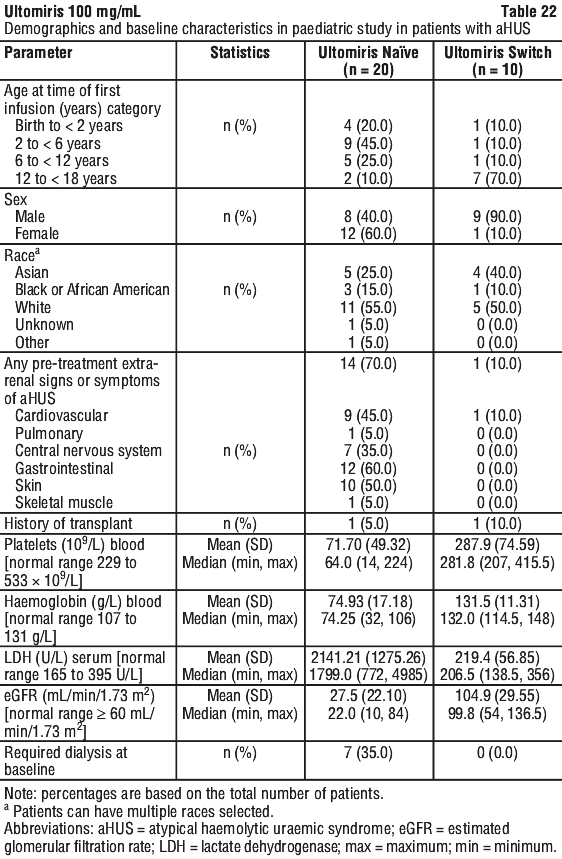
Complete TMA Response was observed in 15 of the 20 naive patients (75.0%) during the 26-week Initial Evaluation Period as shown in Table 23.
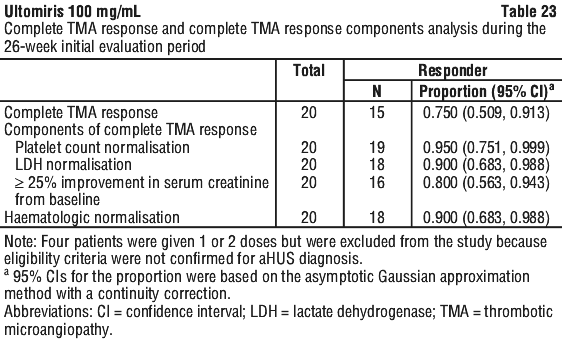
Complete TMA Response was observed in three additional patients during the Extension Period (2 patients on Day 295 and 1 patient on Day 351) resulting in the achievement of Complete TMA response in 18 of 20 paediatric patients (90.0%; 95% CI: 68.3%, 98.8%) through end of study. Individual component response increased to 19 of 20 (95.0%; 95% CI: 75.1%, 99.9%) patients each for platelet count normalisation, LDH normalisation, and in 18 of 20 (90.0%; 95% CI; 68.3%, 98.8%) patients for renal function improvement. The improvement noted in mean eGFR observed by Week 26 was sustained through the end of the study.
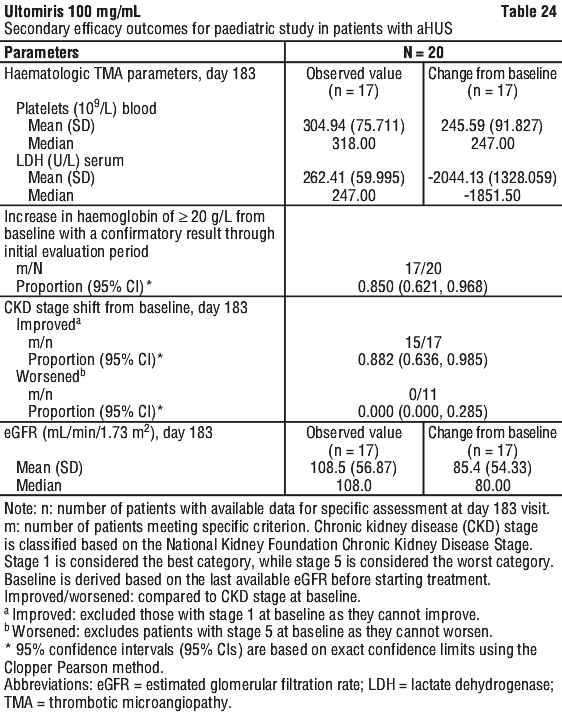
Table 24 summarises the secondary efficacy results for Study ALXN1210-aHUS-312.
All 7 patients who required dialysis at study entry were able to discontinue dialysis; 6 of which had already done so by Day 36. No patient started or re-initiated dialysis during the study. For the 16 patients with available baseline and Week 52 (Day 351) data, 16 patients had improvement in CKD stage compared with baseline. Patients with available data through the end of the study continued to have improvements or no changes in CKD stage.
In Soliris-experienced patients, switching to Ultomiris maintained disease control as evidenced by stable haematologic and renal parameters, with no apparent impact on safety.
Generalised myasthenia gravis (gMG). The efficacy and safety of Ultomiris in adult patients with gMG was assessed in a Phase 3, randomised, double-blind, placebo-controlled, multicenter study (ALXN1210-MG-306) designed to demonstrate Ultomiris superiority over placebo. Patients participating in this study were randomised 1:1 to either receive Ultomiris or placebo for the 26-week Randomised-Controlled Period (RCP), and were subsequently allowed to enter an Open-Label Extension (OLE) Period during which all patients received Ultomiris.
Patients with gMG (diagnosed for at least 6 months) with a positive serologic test for anti-acetylcholine receptor (AChR) antibodies, MGFA (Myasthenia Gravis Foundation of America) clinical classification Class II to IV and Myasthenia Gravis Activities of Daily Living (MG-ADL) total score ≥ 6 were randomised to receive either Ultomiris (N = 86) or placebo (N = 89). Patients on immunosuppressant therapies (ISTs) (corticosteroids, azathioprine, cyclophosphamide, cyclosporine, methotrexate, mycophenolate mofetil, or tacrolimus) were permitted to continue on therapy throughout the course of the study. In addition, rescue therapy (including high dose corticosteroid, PE/PP, or IVIg) was allowed if a patient experienced clinical deterioration, as defined by the study protocol.
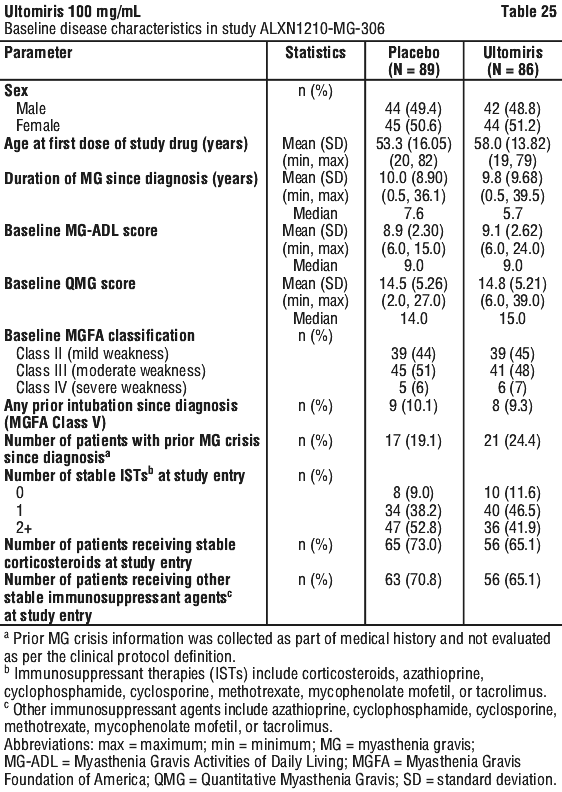
A total of 162 (92.6%) patients completed the 26-week RCP of Study ALXN1210-MG-306. The baseline characteristics of patients were balanced across the 2 treatment groups (see Table 25).
The secondary endpoints, also assessing changes from baseline to Week 26, included the change in the Quantitative Myasthenia Gravis (QMG) total score (QMG - a validated clinician-reported assessment of muscle weakness in gMG), the proportion of patients with improvements of at least 5 and 3 points in the QMG and MG-ADL total scores, respectively, as well as changes in quality of life assessments.
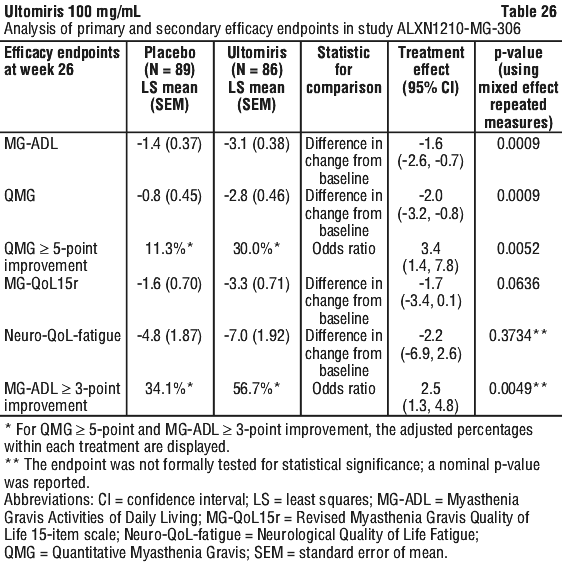
Ultomiris demonstrated a statistically significant change in the primary endpoint, change in MG ADL total score from baseline to Week 26, as compared to placebo. Primary and secondary endpoint results are presented in Table 26.
The treatment effect of Ultomiris on MG-ADL was rapid, with an improvement demonstrated as early as Week 1 (p = 0.0265) and sustained through Week 26. The efficacy of Ultomiris was consistent overall across pre-specified subgroups (sex, age, body weight, region, immunosuppressant therapy use at baseline, and MGFA clinical classification).
In patients treated with Ultomiris, improvements were observed in all domain scores of the MG-ADL and in the ocular, bulbar, and limb domain scores of the QMG.
Other efficacy results. Overall, 25.6% of patients treated with Ultomiris achieved minimal manifestation status as per the MGFA post-intervention status at Week 26 compared to 9.9% of patients treated with placebo.
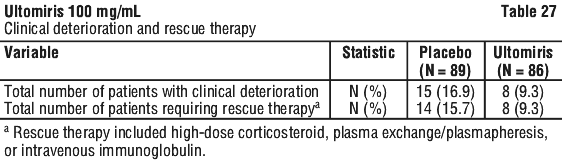
Table 27 presents an overview of the patients with clinical deterioration and patients requiring rescue therapy over the 26-week RCP.

Neuromyelitis optica spectrum disorder (NMOSD). Study in adult patients with NMOSD. The efficacy of Ultomiris in adult patients with anti-AQP4 antibody-positive NMOSD was assessed in a global, open-label clinical study (ALXN1210-NMO-307).
Study ALXN1210-NMO-307 enrolled 58 adult patients with NMOSD who had a positive serologic test for anti-AQP4 antibodies, at least 1 relapse in the last 12 months prior to the screening period, and an expanded disability status scale (EDSS) score of ≤ 7. Prior treatment with immunosuppressant therapies (ISTs) was not required for enrolment and 51.7% of patients were on Ultomiris monotherapy. Patients on selected ISTs (i.e. corticosteroids, azathioprine, mycophenolate mofetil, tacrolimus) were permitted to continue on therapy in combination with Ultomiris, with a requirement for stable dosing until they reached Week 106 in the study. In addition, acute therapy for relapse treatment (including high-dose corticosteroids, PE/PP, and IVIg) was allowed if a patient experienced a relapse during the study.
Patients included in the study had a mean age of 47.4 years (ranging from 18 to 74 years) and most of them were female (90%). Median age at NMOSD initial clinical presentation was of 42.5 years, ranging from 16 to 73 years. Baseline disease characteristics are shown in Table 28.
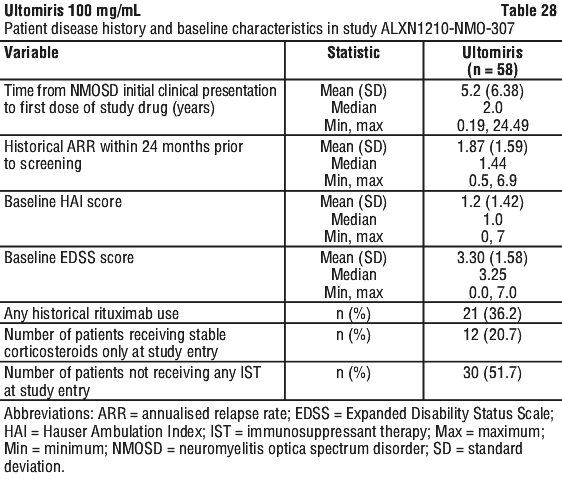
A pre-specified indirect comparison of the Ultomiris arm of the study with an external placebo arm (placebo arm of the phase 3 Study ECU-NMO-301) showed a relative relapse risk reduction of 98.6% compared to placebo (hazard ratio: 0.014; p < 0.0001).
All Ultomiris-treated patients remained relapse free over the median follow-up of 90.93 weeks. Ultomiris-treated patients experienced consistent relapse-free primary endpoint result with or without concomitant IST treatment.
Ultomiris-treated patients had an adjudicated on-trial annualized relapse rate (ARR) that was statistically significantly lower than the predicted ARR of 0.25 (1 adjudicated on-trial relapse per 4 patient -years) (p < 0.0001). The 0.25 comparator rate was chosen to represent a conservative ARR that may be experienced in the NMOSD patient population.
Of the 58 Ultomiris-treated patients, 96.6% and 89.7% did not experience clinically important worsening in Hauser Ambulation index score and expanded disability status scale during the primary treatment period, respectively.
Ultomiris has not been studied for the acute treatment of relapses in NMOSD patients.
5.2 Pharmacokinetic Properties
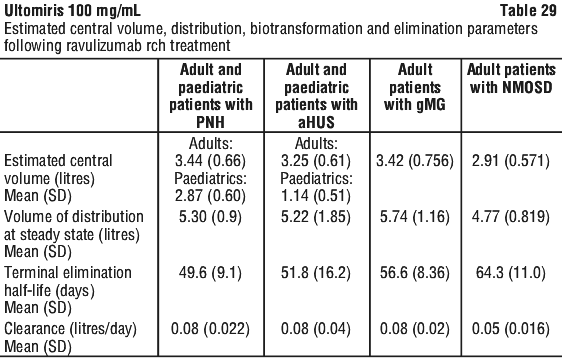
A linear, 2-compartment PK model was developed that adequately described the observed ravulizumab rch PK following IV administration. The estimated mean (SD) clearance, central volume, volume at steady state and terminal elimination half-life following multiple dosing of ravulizumab rch in adult and paediatric patients with PNH or aHUS and in adult patients with gMG and NMOSD are provided in Table 29.
In patients with PNH, aHUS, gMG or NMOSD pharmacodynamic activity correlates directly with ravulizumab rch serum concentrations above the target exposure level results in free C5 levels < 0.5 microgram/mL, achieving immediate, complete and sustained blockade of terminal complement inhibition in all patients.
Absorption. Because ravulizumab rch administration is via an IV infusion and the dosage form is a solution, 100% of the administered dose is considered bioavailable. The time to maximum observed concentration (tmax) is expected to be at the end of infusion (EOI) or soon after EOI. Over the studied dose and regimen range, ravulizumab rch exhibited dose proportional and time linear pharmacokinetics (PK).
Distribution. The mean (standard deviation [SD]), central volume and volume of distribution at steady state in adult and paediatric patients with PNH, aHUS, or adult patients with gMG and NMOSD treated with ravulizumab rch are presented in Table 29.
Metabolism and excretion. As an IgG monoclonal antibody, ravulizumab rch is expected to be metabolised in the same manner as any endogenous IgG (degraded into small peptides and amino acids via catabolic pathways), and is subject to similar elimination. Ravulizumab rch contains only natural occurring amino acids and has no known active metabolites. The mean (SD) values for terminal elimination half-life and clearance of ravulizumab rch in patients with PNH, aHUS, gMG or NMOSD treated with ravulizumab rch are presented in Table 29.
Body weight was a significant covariate on the pharmacokinetics of ravulizumab rch.
Special populations. No formal trial of the effect of sex, race, age (elderly), hepatic or renal impairment on the pharmacokinetics of ravulizumab rch was conducted. However, based on population-PK assessment, no impact of sex, age, race and hepatic or renal function on ravulizumab rch PK was identified in the studied healthy volunteer subjects and patients with PNH, aHUS, gMG, or NMOSD and as a result, no dosing adjustment is considered necessary.
The pharmacokinetics of ravulizumab rch have been studied in aHUS patients with a range of renal impairment, including patients receiving dialysis. There have been no observed differences in pharmacokinetic parameters noted in these sub-populations, including patients with proteinuria.
5.3 Preclinical Safety Data
Genotoxicity. No studies have been conducted to assess the genotoxic potential of ravulizumab rch.
Carcinogenicity. No studies have been conducted to assess the carcinogenic potential of ravulizumab rch.
6 Pharmaceutical Particulars
6.1 List of Excipients
Monobasic sodium phosphate, dibasic sodium phosphate, polysorbate 80, L-arginine, sucrose, water for injections.
Ultomiris 100 mg/mL contains 4.6 mg sodium per 3 mL vial or 16.8 mg sodium per 11 mL vial. This should be taken into consideration by patients on a controlled sodium diet.
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products.
Reconstitution and dilution should only use 0.9% sodium chloride, solution for injection as diluent.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Ultomiris vials must be stored under refrigerated conditions at 2°C - 8°C.
After dilution, the medicinal product should be used immediately. However, chemical and physical stability of the diluted product have been demonstrated for up to 24 hours at 2°C-8°C and up to 4 hours at room temperature.
Vials must not be frozen or shaken.
Keep the vial in the outer carton to protect from light.
6.5 Nature and Contents of Container
Type I glass vial with a stopper and a seal.
Pack size of one vial.
6.6 Special Precautions for Disposal
Product is for single use in one patient only. Discard any residue.
In Australia, any unused medicine or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
Chemical structure.
7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.
Date of First Approval
23 March 2021
Date of Revision
14 January 2026
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.