Vimizim
Brand Information
| Brand name | Vimizim |
| Active ingredient | Elosulfase alfa |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Vimizim.
Summary CMI
Vimizim®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I using Vimizim?
Vimizim contains the active ingredient elosulfase alfa (rch). Vimizim is used to treat patients with Mucopolysaccharidosis Type IVA (MPS IVA). For more information, see Section 1. Why am I using Vimizim? in the full CMI.
2. What should I know before I use Vimizim?
Do not use if you have ever had an allergic reaction to Vimizim or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use Vimizim? in the full CMI.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines that you buy without a prescription from your pharmacy, supermarket or health food. For more information, see Section 3. What if I am taking other medicines? in the full CMI.
4. How am I given Vimizim?
- The dose you receive is based on your body weight. The recommended dose regimen is 2 mg/kg body weight administered once every week through a drip into a vein (by intravenous infusion).
- Your doctor or nurse will administer Vimizim to you.
- Each infusion will take approximately 4 hours, and your doctor will decide how long you will receive Vimizim for.
More instructions can be found in Section 4. How am I given Vimizim? in the full CMI.
5. What should I know while being treated with Vimizim?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while being treated with Vimizim? in the full CMI.
6. Are there any side effects?
All medicines can have side effects. If they do occur, they are usually minor and temporary. Do not be alarmed by this list. You may not experience any of them.
The most common side effects include ear pain, feeling nervous or jittery, headache, dizziness, flushing, tingling or prickling of the skin, drowsiness, mouth and throat pain, shortness of breath, vomiting, nausea, diarrhoea, abdominal pain, hives, neck or muscle pain, fever, chills, infusion site pain, chest discomfort and cloudiness in the eye. The most serious side effect is life-threatening allergic reaction.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
Vimizim®
Active ingredient: elosulfase alfa (rch)
Consumer Medicine Information (CMI)
This leaflet provides important information about using Vimizim. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using Vimizim.
Where to find information in this leaflet:
1. Why am I using Vimizim?
2. What should I know before I use Vimizim?
3. What if I am taking other medicines?
4. How am I given Vimizim?
5. What should I know while being treated with Vimizim?
6. Are there any side effects?
7. Product details
1. Why am I using Vimizim?
Vimizim contains the active ingredient elosulfase alfa, which belongs to a group of medicines known as enzyme replacement therapies.
Elosulfase alfa is a recombinant version of a human enzyme produced by genetic engineering in Chinese Hamster Ovary (CHO) cells. It works by replacing the natural enzyme in patients with Mucopolysaccharidosis Type IVA (MPS IVA, also known as Morquio A Syndrome).
Vimizim is used to treat patients with MPS IVA. People with MPS IVA have either a low level, or reduced activity, of an enzyme called N-acetylgalactosamine-6-sulfatase (or GALNS), which breaks down specific substances (for example, keratan sulfate) in the body. As a result, these specific substances do not get broken down and processed by the body as they should. They accumulate in many tissues in the body, which causes the symptoms of MPS IVA.
Treatment with Vimizim has shown improvement in walking ability and reduction of the levels of keratan sulfate.
2. What should I know before I use Vimizim?
Warnings
You must not receive Vimizim if:
- you have experienced severe or life-threatening allergic reactions to elosulfase alfa, or any of the ingredients listed at the end of this leaflet, and your doctor is not able to control these reactions withmedicines or other measures such as slowing the rate of the infusion or temporarily stopping the infusion.
Always check the ingredients to make sure you can use this medicine.
Your doctor will tell you if you can have Vimizim or not.
Check with your doctor if you:
- have any other medical conditions, especially the following:
- a severe allergic reaction to elosulfase alfa or any of the other ingredients of Vimizim
- any severe side effects with previous Vimizim treatment
- fever
- any respiratory conditions including sleep apnoea (a condition where you temporarily stop breathing during your sleep) - are on a controlled sodium diet, as each 5 mL vial of Vimizim contains 8 mg of sodium
- have an intolerance to certain sugars, as each 5 mL vial of Vimizim contains 100 mg of sorbitol
- have allergies to any other medicines, foods, preservatives or dyes.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
Vimizim has not been studied in pregnant patients and should not be given during pregnancy unless clearly necessary.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
It is not known if Vimizim passes into breast milk. Discuss with your doctor the risks and benefits of continuing to take Vimizim while breastfeeding.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket, naturopath or health food shop.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect Vimizim.
4. How am I given Vimizim?
How Vimizim is given
Your doctor or nurse will administer Vimizim to you as a drip into a vein (by intravenous infusion). The medicine has to be diluted before being given and should not be mixed with other products.
Your doctor or nurse may give you some medicines before your treatment to reduce allergic reactions and you may also be given medicines to help control any fever.Your doctor will decide how long you will receive Vimizim for.
How much is given
The dose you receive is based on your body weight. The recommended dose regimen is 2 mg/kg body weight administered once every week through a drip into a vein (by intravenous infusion).
Each infusion will take approximately 4 hours.
If you miss a dose
If you miss a dose, talk to your doctor or nurse and arrange another visit as soon as possible.
If you take too much Vimizim
Vimizim is administered under the supervision of a health care professional. He or she will check that the correct dose has been given and act accordingly if necessary.
5. What should I know while being treated with Vimizim?
Things you should do
Your condition MPS IVA can cause pressure on the spinal cord. Call your doctor straight away if you experience:
- back pain
- numbness or loss of feeling in parts of your body
- any bowel or bladder problems
When you are treated with Vimizim, you may develop infusion reactions. An infusion reaction is any side effect, including an allergic reaction, occurring during the infusion or within a day following infusion. Call your doctor immediately if you think you are experiencing such a reaction.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
- cough
- throat tightness
- bluish colour of skin
- abnormally low blood pressure
- chest discomfort
- gastrointestinal symptoms, such as nausea, stomach ache, dry heaves, and vomiting.
If you have an allergic reaction, your doctor may slow down, or stop your infusion. Your doctor may also give you additional medicines to manage any allergic reaction. Your doctor will decide when you can restart Vimizim treatment.
- Tell your doctor if you have sleep apnoea or use a CPAP machine. You may need to have it available during infusions.
Keep all appointments with your doctor and always discuss anything that worries you during or after treatment with Vimizim.
If you become pregnant while you are treated with Vimizim, tell your doctor immediately.
Remind any doctor, nurse, dentist or pharmacist you visit that you are using Vimizim.
Things you should not do
Do not stop your treatment visits for Vimizim unless you have spoken to your doctor.
Your condition may worsen if you stop receiving Vimizim.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how Vimizim affects you.
Vimizim may cause dizziness in some people. If you feel dizzy during or after your infusion, do not drive or operate machinery.
Looking after your medicine
Vimizim must be stored in a refrigerator at 2°C to 8°C, but it must not be frozen. The vial should be kept in the carton to protect it from light. Your doctor or pharmacist is responsible for storing Vimizim.
When to discard your medicine
Each vial of Vimizim should be used once only. The doctor or nurse will discard any unused portion and dispose of used vial.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
Infections related:
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
Allergy related:
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What Vimizim contains
| Active ingredient (main ingredient) | 5 mg elosulfase alfa in each vial (1 mg/mL) |
| Other ingredients (inactive ingredients) |
|
Do not take this medicine if you are allergic to any of these ingredients.
What Vimizim looks like
Vimizim is a clear, colourless to pale yellow concentrated solution for injection in a clear glass vial containing 5 mL of solution. Each pack contains 1 vial. (Aust R 215523).
Who distributes Vimizim
Vimizim is supplied in Australia by:
BioMarin Pharmaceutical Australia Pty Ltd
119 Willoughby Road
Crows Nest, NSW 2065
Telephone (02) 8520 3255
Vimizim is supplied in New Zealand by:
Pharmacy Retailing (NZ) Limited t/a Healthcare Logistics
58 Richard Pearse Drive
Airport Oaks 2022
Auckland
Telephone (09) 918 5100
For enquiries about Vimizim, contact medinfoasia@bmrn.com or call BioMarin:
Australia: 1800 387 876
New Zealand: 0800 882 012
To report adverse events, contact drugsafety@bmrn.com or call BioMarin:
Australia: 1800 387 876
New Zealand: 0800 882 012
This leaflet was prepared in December 2025.
® Registered trademark of BioMarin Pharmaceutical Inc., USA
Brand Information
| Brand name | Vimizim |
| Active ingredient | Elosulfase alfa |
| Schedule | S4 |
MIMS Revision Date: 01 March 2019
1 Name of Medicine
Elosulfase alfa (rch).
2 Qualitative and Quantitative Composition
Each mL of solution contains 1 mg elosulfase alfa. *Each vial of approximately 5 mL extractable solution contains 5 mg elosulfase alfa.
*Elosulfase alfa is a purified human enzyme produced by recombinant DNA technology in a Chinese hamster ovary cell line.
Human N-acetylgalactosamine-6-sulfatase (EC 3.1.6.4) is a lysosomal enzyme that hydrolyses sulfate from either galactose-6-sulfate or N-acetyl-galactosamine-6-sulfate on the non-reducing ends of the glycosaminoglycans keratan sulfate (KS) and chondroitin sulfate.
Elosulfase alfa is a soluble dimeric protein, and each monomer contains 496 amino acids with an approximate molecular mass of 55 kDa for the peptide chain. The oligosaccharides present at the two consensus N-linked glycosylation sites contain mannose-6-phosphate (M6P). M6P is recognised by a receptor at the cell surface and is crucial for efficient cellular uptake of the protein to the lysosome. Elosulfase alfa has a specific activity of 2.5 to 6.0 units/mg. One activity unit is defined as the amount of the enzyme required to convert 1 micromole of sulfated monosaccharide substrate D-galactopyranoside-6-sulfate (Gal-6S) to de-sulfated-galactose (Gal) and free sulfate per minute at 37°C.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Concentrated solution for injection.
A sterile, nonpyrogenic, colourless to pale yellow and clear to slightly opalescent solution with a pH between 5.0 to 5.8 that must be diluted with sodium chloride 9 mg/mL (0.9%) solution for injection prior to administration.
4 Clinical Particulars
4.1 Therapeutic Indications
Vimizim is indicated for the treatment of mucopolysaccharidosis type IVA (MPS IVA; Morquio A syndrome).
4.2 Dose and Method of Administration
Vimizim treatment should be supervised by a physician or healthcare provider experienced in the management of patients with MPS IVA or other inherited metabolic diseases. Administration of Vimizim should be carried out by an appropriately trained healthcare professional with the ability to manage medical emergencies. Home administration under the supervision of a healthcare professional trained in recognising and medically managing serious infusion related reactions under the direction of a practicing physician may be considered only for patients who are tolerating their infusions well.
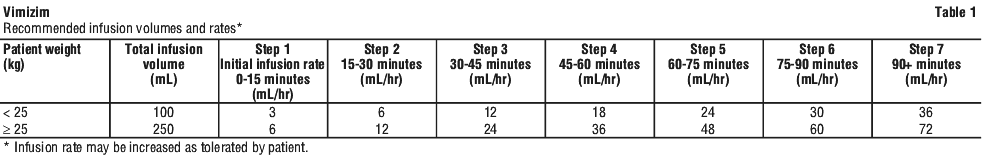
Dose. The recommended dosage for Vimizim is 2 mg/kg of bodyweight administered once a week. The total volume of the infusion should be delivered over approximately 4 hours (hr) (see Table 1).
Pre-treatment with antihistamines with or without antipyretics is recommended 30-60 minutes prior to start of infusion (see Section 4.4 Special Warnings and Precautions for Use, Infusion reactions).
Special populations. Elderly patients. No alternative dosage regimen can be recommended for elderly patients (see Section 4.4 Special Warnings and Precautions for Use, Use in the elderly).
Paediatric population. Dosage and administration are the same as in adults (see Section 4.8 Adverse Effects (Undesirable Effects); Section 5.1 Pharmacodynamic Properties).
Method of administration. Vimizim must be diluted with sodium chloride 9 mg/mL (0.9%) solution for injection, to a final volume of 100 mL or 250 mL based on the patient's weight, prior to infusion, and delivered via intravenous infusion.
The final volume is based on the patient's weight as follows:
for patients who weigh less than 25 kg, the final volume should be 100 mL;
for patients who weigh 25 kg or more, the final volume should be 250 mL.
When diluted in 100 mL, the initial infusion rate should be 3 mL/hr. The infusion rate may be increased as tolerated, every 15 minutes as follows: first increase the rate to 6 mL/hr, then increase the rate every 15 minutes by 6 mL/hr increments until a maximum rate of 36 mL/hr is reached.
When diluted in 250 mL, the initial infusion rate should be 6 mL/hr. The infusion rate may be increased as tolerated, every 15 minutes as follows: first increase the rate to 12 mL/hr, then increase the rate every 15 minutes by 12 mL/hr increments until a maximum rate of 72 mL/hr is reached.
Preparation of the Vimizim infusion (aseptic technique is to be used). 1. Determine the number of vials to be diluted based on the individual patient's weight and the recommended dose of 2 mg/kg, using the following calculation:
patient weight (kg) multiplied by 2 mg per kg = Patient dose (mg);
patient dose (mg) divided by (1 mg/mL concentrate of Vimizim) = Total number of mL of Vimizim;
total amount (mL) Vimizim divided by 5 mL per vial = Total number of vials.
2. Round up to the next whole vial. Remove the appropriate number of vials from the refrigerator. Do not heat or microwave vials. Do not shake vials.
3. Obtain an infusion bag containing sodium chloride 9 mg/mL (0.9%) solution for injection suitable for intravenous administration. The total volume of the infusion is determined by the patient's body weight.
Patients weighing less than 25 kg should receive a total volume of 100 mL.
Patients weighing 25 kg or more should receive a total volume of 250 mL.
4. Before withdrawing Vimizim from the vial, visually inspect each vial for particulate matter and discolouration. Because this is a protein solution, slight flocculation (thin translucent fibres) may occur. The Vimizim solution should be clear to slightly opalescent and colourless to pale yellow. Do not use if the solution is discoloured or if there is particulate matter in the solution.
5. Withdraw and discard a volume of the sodium chloride 9 mg/mL (0.9%) solution for injection from the infusion bag, equal to the volume of Vimizim concentrate to be added.
6. Slowly withdraw the calculated volume of Vimizim from the appropriate number of vials using caution to avoid excessive agitation.
7. Slowly add Vimizim to the infusion bag using care to avoid agitation.
8. Gently rotate the infusion bag to ensure proper distribution of Vimizim. Do not shake the solution.
9. Administer the diluted Vimizim solution to patients using a low-protein-binding infusion set equipped with a low protein binding 0.2 micrometer in-line filter.
Vimizim does not contain preservatives; therefore the product should be used immediately after dilution.
Vimizim should not be infused with other products in the infusion bag as compatibility with other products has not been established.
4.3 Contraindications
Severe or life-threatening hypersensitivity to the active substance or to any of the excipients, if hypersensitivity is not controllable.
4.4 Special Warnings and Precautions for Use
Anaphylaxis and severe allergic reaction. Anaphylaxis and severe allergic reactions have been reported in clinical studies. Therefore, appropriate medical support must be readily available when Vimizim is administered. If these reactions occur, immediately stop the infusion of Vimizim and initiate appropriate medical treatment. The current medical standards for emergency treatment are to be followed. For patients who have experienced severe allergic reactions during infusion with Vimizim, caution should be exercised upon re-challenge. Pretreatment with corticosteroids and/or reduction in the infusion rate in addition to antihistamines and antipyretics should be considered for subsequent infusions.
In clinical trials, anaphylaxis was reported as early as 30 minutes from the start of infusion and up to three hours after infusion. Anaphylaxis occurred as late into treatment as the 47th infusion. The signs and symptoms of anaphylaxis include cough, erythema, throat tightness, urticaria, flushing, cyanosis, hypotension, rash, dyspnoea, chest discomfort, and gastrointestinal symptoms (e.g. nausea, abdominal pain, retching, and vomiting) in conjunction with urticaria (see Section 4.8 Adverse Effects (Undesirable Effects)).
Hypersensitivity reactions reported in clinical trials occurred as early as 30 minutes from the start of infusion but as late as six days after infusion. Frequent symptoms of hypersensitivity reactions (occurring in more than 2 patients) included anaphylactic reactions, urticaria, peripheral oedema, cough, dyspnoea, and flushing (see Section 4.8 Adverse Effects (Undesirable Effects)).
Observe patients closely for an appropriate period of time after administration of Vimizim, taking into account the time to onset of anaphylaxis seen in premarketing clinical trials. Inform patients of the signs and symptoms of anaphylaxis, and instruct them to seek immediate medical care should signs and symptoms occur.
Infusion reactions. In clinical trials, 96% of patients treated with Vimizim experienced infusion reactions (IRs). IRs may include allergic reactions. In patients who experienced IRs, subsequent infusions were managed with slower infusion rates, treatment with additional prophylactic antihistamines and, in the event of a more severe reaction, treatment with prophylactic corticosteroids. Thirty-five patients (15.2%) discontinued at least one infusion due to an IR. Sixty (0.66%) of the 9,126 infusions administered in the clinical trials were discontinued due to an IR. 17.3% of patients had an infusion reaction requiring medical intervention. In 13 out of 231 patients and less than 1% of the total infusions, the infusion was discontinued and medical intervention was required.
Because of the potential for IRs with Vimizim, patients should receive antihistamines with or without antipyretics prior to infusion. Management of IRs should be based on the severity of the reaction and include slowing or temporary interruption of the infusion and/or administration of additional antihistamines, antipyretics, and/or corticosteroids. If severe IRs occur, immediately stop the infusion of Vimizim and initiate appropriate treatment. In case of a recurrent IR or re-challenge after a single severe IR, pre-treatment should be considered (antihistamines and antipyretics and/or corticosteroids) and a reduction of the infusion rate to 50% - 25% of the rate at which the previous reaction occurred. The risks and benefits of re-administering Vimizim following a severe reaction should be considered and patients should be closely monitored by the treating physician (see Section 4.2 Dose and Method of Administration).
Spinal/cervical cord compression. Spinal/cervical cord compression (SCC) is a known and serious complication of MPS IVA and may occur as part of the natural history of the disease. In clinical trials, SCC was observed both in patients receiving Vimizim and patients receiving placebo. Patients with MPS IVA should be monitored for signs and symptoms of SCC (including back pain, paralysis of limbs below the level of compression, urinary and fecal incontinence) and given appropriate clinical care.
Acute respiratory complications associated with administration. Patients with acute febrile or respiratory illness at the time of Vimizim infusion may be at higher risk of life-threatening complications from hypersensitivity reactions. Careful consideration should be given to the patient's clinical status prior to administration of Vimizim and consider delaying the Vimizim infusion.
Sleep apnoea is common in MPS IVA patients. Evaluation of airway patency should be considered prior to initiation of treatment with Vimizim. Patients using supplemental oxygen or continuous positive airway pressure (CPAP) during sleep should have these treatments readily available during infusion in the event of an acute reaction, or extreme drowsiness/sleep induced by antihistamine use.
Sodium restricted diet. This medicinal product contains 8 mg sodium per vial and is administered in sodium chloride 9 mg/mL (0.9%) solution for injection (see Section 4.2 Dose and Method of Administration). This should be taken into consideration for patients on a controlled sodium diet.
Sorbitol. Patients with rare hereditary problems of fructose intolerance should not take this medicine.
Use in the elderly. The safety and efficacy of Vimizim in patients older than 65 years have not been established and it is not known whether they respond differently from younger patients.
Paediatric use. The majority of patients treated with Vimizim in clinical trials were in the paediatric age range (53% aged 5 to 11 years, 27% aged 12-17 years). Patients < 5 years of age were not included in the pivotal study (MOR-004). In an open-label trial (MOR-007), 15 paediatric patients with MPS IVA under the age of 5 years (9 months to < 5 years) received 2 mg/kg of Vimizim once a week for 52 weeks. Patients continued a long-term follow-up observational study. The mean duration of dosing was 125.3 weeks and ranged from 53.0 to 200.9 weeks. Efficacy assessment by a 6 minute walk test was not conducted due to the young age of these patients. However, in the MOR-007 study, patients showed a reduction of urinary KS; and safety results in patients under the age of 5 years are consistent with results observed in patients 5 to 57 years old (see Section 4.8 Adverse Effects (Undesirable Effects); Section 5.1 Pharmacodynamic Properties, Clinical trials).
Effects on laboratory tests. No data available.
4.5 Interactions with Other Medicines and Other Forms of Interactions
No interaction studies have been performed.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. A combined fertility/embryo-fetal development study was conducted in male and female rats administered intravenous elosulfase alfa up to 20 mg/kg/day with diphenhydramine (DPH) 10 mg/kg intraperitoneal (IP), prior to mating and during the cohabitation period. Dosing of females continued through Gestation Day 20. At systemic exposures up to around 200-400 times the human value, based on AUC, there was no evidence of impaired fertility or reproductive performance.
Use in pregnancy. (Category B3)
There are no adequate and well controlled studies in pregnant women receiving Vimizim.
Reproduction studies were performed in rats receiving up to 20 mg/kg/day (AUC exposure ratio around 200) elosulfase alfa with DPH (10 mg/kg, IP) from premating through Gestation Day 20 and rabbits receiving up to 10 mg/kg/day (AUC exposure ratio greater than 30) elosulfase alfa from Gestation Day 7 through Gestation Day 20. There were no elosulfase alfa-related effects on embryo-fetal development, and no increased incidence of fetal gross external, soft tissue or skeletal alterations in rats or rabbits. However, administration of elosulfase alfa to rats at 6 or 20 mg/kg/day (predicted AUC exposure ratios of around 40 and 200, respectively) from Gestation Day 7 through Lactation Day 20, produced significant increases in perinatal pup mortality. As a precautionary measure, it is preferable to avoid the use of Vimizim during pregnancy, unless clearly necessary.
Use in lactation. Data from rats have shown excretion of Vimizim in milk. It is not known whether Vimizim is excreted in human breast milk, therefore a decision on whether to continue/discontinue breast-feeding or to continue/discontinue therapy with Vimizim should be made, taking into account the benefit of breast-feeding to the child and the benefit of Vimizim therapy to the woman.
4.7 Effects on Ability to Drive and Use Machines
No studies of Vimizim effects on the ability to drive and use machines have been performed. Dizziness was reported during Vimizim infusions; if dizziness occurs after the infusion, the ability to drive and use machines may be affected.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile. The assessment of adverse reactions is based on the exposure of 176 patients with MPS IVA, ages 5 to 57 years old to 2 mg/kg Vimizim once a week (n=58, mean duration 23.6 ± 3.03 weeks), 2 mg/kg Vimizim once every other week (n=59, mean duration 24.0 ± 0.19 weeks), or placebo (n=59) in a randomised, double-blind, placebo-controlled trial (MOR-004).
The majority of related adverse events in clinical trials were IRs (reported in 96% of patients treated with Vimizim), which are defined as reactions occurring after initiation of infusion until the end of the day following the infusion. Serious IRs were observed in clinical trials and included anaphylaxis, hypersensitivity and vomiting (see Section 4.4 Special Warnings and Precautions for Use, Anaphylaxis and severe allergic reaction and Infusion reactions). Forty-four of 235 (18.7%) patients experienced hypersensitivity reactions, and 18 of 235 (7.7%) patients treated with Vimizim experienced signs and symptoms consistent with a clinical diagnosis of anaphylaxis based on USA National Institute of Allergy and Infectious Disease and Food Allergy and Anaphylaxis Network (NIAID/FAAN) criteria. These 18 patients experienced 26 anaphylactic reactions out of > 11,000 infusions (0.24%) (see Section 4.4 Special Warnings and Precautions for Use, Anaphylaxis and severe allergic reaction).
The most common symptoms of IRs (occurring in ≥ 10% of patients treated with Vimizim and ≥ 5% more when compared to placebo) were headache, nausea, vomiting, pyrexia, chills and abdominal pain. IRs were generally mild or moderate, and the frequency was higher during the first 12 weeks of treatment and tended to occur less frequently with time.
Tabulated list of adverse reactions. The most common adverse events reported in the pivotal trial with an incidence of ≥ 5% more in patients treated with Vimizim (2 mg/kg per week) than in placebo treated patients, regardless of causality, are listed in Table 2.
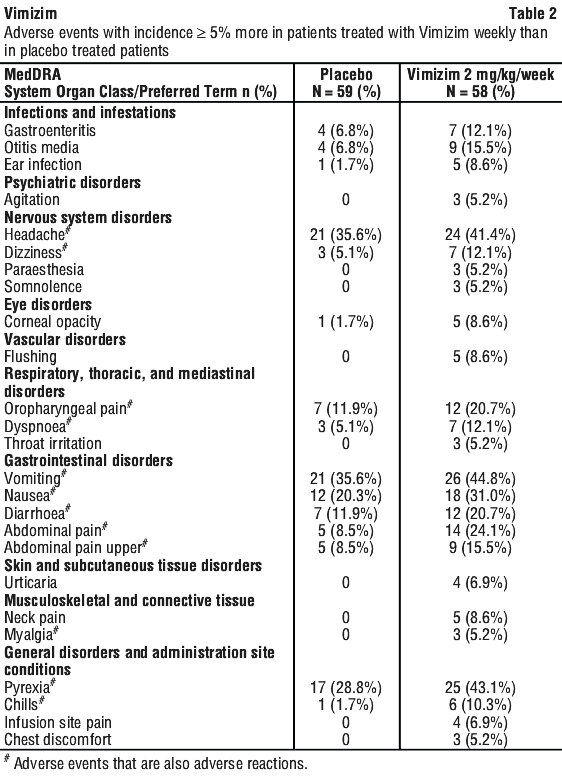
Other adverse reactions not listed in the table. In the placebo controlled study, hypersensitivity was reported in 7 (5.9%) patients receiving Vimizim in both 2 mg/kg per week and 2 mg/kg every other week groups compared to 1 (1.7%) patient in the placebo group. Severe hypersensitivity was reported in one of these patients in the 2 mg/kg Vimizim per week group. Anaphylactic reaction was also reported in one patient in the 2 mg/kg Vimizim every other week group 6 weeks after initiating study medication (see Section 4.4 Special Warnings and Precautions for Use, Anaphylaxis and severe allergic reaction).
The nature and severity of adverse reactions observed in other clinical trials were similar to the adverse reactions observed in the pivotal trial. One patient discontinued during open label treatment with Vimizim due to an adverse event.
Description of adverse reactions. Immunogenicity. All patients treated with Vimizim developed sustained anti-drug antibodies. Approximately 80% of patients developed neutralising antibodies capable of inhibiting the drug from binding to the cation-independent mannose-6-phosphate receptor. Sustained improvements in efficacy measures and reductions in urine keratan sulfate (KS) over time were observed across trials, despite the presence of anti-drug antibodies. No correlations were found between higher antibody titres or neutralising antibody positivity and reductions in efficacy measurements or occurrence of anaphylaxis or other hypersensitivity reactions. Immunoglobulin E (IgE) antibodies against Vimizim were detected in ≤ 10% of treated patients and have not consistently been related to anaphylaxis or other hypersensitivity reactions and/or treatment withdrawal.
Paediatric population. Adverse reactions observed in paediatric patients < 5 years of age. In the MOR-007 clinical trial, 15 paediatric patients < 5 years of age (range 9 months to 4.9 years) were treated with Vimizim 2 mg/kg once per week over 52 weeks. The most commonly reported adverse reactions were pyrexia (100%) and vomiting (80%). Other adverse reactions occurring in 2 or more patients were diarrhoea, abdominal pain, abdominal pain upper, oropharyngeal pain, headache, hypersensitivity, and nausea. One serious adverse reaction of hypersensitivity was reported during this period.
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
4.9 Overdose
There is no experience of overdose in clinical trials.
For information on the management of overdose, contact the Poison Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action. Mucopolysaccharidoses (MPS) comprise a group of lysosomal storage disorders caused by the deficiency of specific lysosomal enzymes required for the catabolism of glycosaminoglycans (GAG). MPS IVA is characterised by the absence or marked reduction in N-acetylgalactosamine-6-sulfatase activity. The sulfatase activity deficiency results in the accumulation of the GAG substrates, KS and chondroitin 6 sulfate (C6S), in the lysosomal compartment of cells throughout the body. The accumulation leads to widespread cellular, tissue, and organ dysfunction. Vimizim is intended to provide the exogenous enzyme N-acetylgalactosamine-6-sulfatase that will be taken up into the lysosomes and increase the catabolism of the GAGs KS and C6S. Cell uptake of Vimizim into lysosomes is through cation independent mannose-6-phosphate receptors mediated internalisation leading to restored GALNS activity and clearance of KS. Extracellular KS was not affected by Vimizim treatment, verifying that Vimizim activity was restricted to the lysosome.
Pharmacodynamic effects. The pharmacodynamic effect of Vimizim was assessed by reductions in urinary KS levels. The relationship of urinary KS to other measures of clinical response has not been established (see Section 5.1 Pharmacodynamic Properties, Clinical trials). No association was observed between antibody development and urinary KS levels.
Clinical trials. Clinical trials performed with Vimizim assessed the impact of treatment on the systemic manifestations of MPS IVA in various domains including endurance, respiratory function, growth velocity, and mobility, as well as urine KS.
A total of 244 patients with MPS IVA were enrolled and exposed to Vimizim in six clinical trials.
The safety and efficacy of Vimizim was assessed in a randomised, double-blind, placebo-controlled, Phase 3 clinical trial (MOR-004) of 176 patients with MPS IVA, ranging in age from 5 to 57 years. The majority of the patients (82%) presented with a medical history of musculoskeletal conditions, which includes knee deformity (52%), kyphosis (31%), hip dysplasia (22%), prior spinal fusion surgery (22%) and arthralgia (20%). Patients also presented with short stature and impaired endurance. Patients who could walk more than 30 metres (m) but less than 325 m in a 6 Minute Walk Test (MWT) at baseline were enrolled in the trial.
Patients received Vimizim 2 mg/kg every week (n=58) or 2 mg/kg every other week (n=59), or placebo (n=59) for a total of 24 weeks. All patients were treated with antihistamines prior to each infusion.
The primary endpoint was the change from baseline in the 6-MWT distance compared to placebo at Week 24. The secondary endpoints were the change from baseline in the 3 Minute Stair Climb Test (MSCT) and urine KS levels at Week 24.
A total of 173 patients subsequently enrolled in an extension trial (MOR-005) in which patients received 2 mg/kg of Vimizim every week or 2 mg/kg every other week, and then all were switched to 2 mg/kg every week upon availability of the Week 24 results.
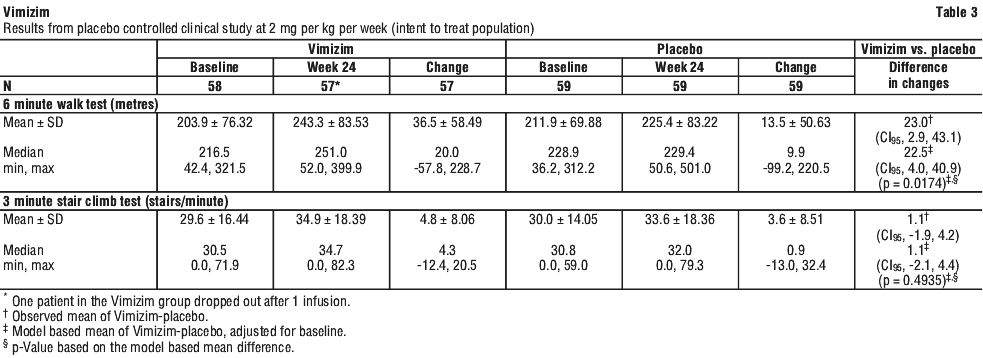
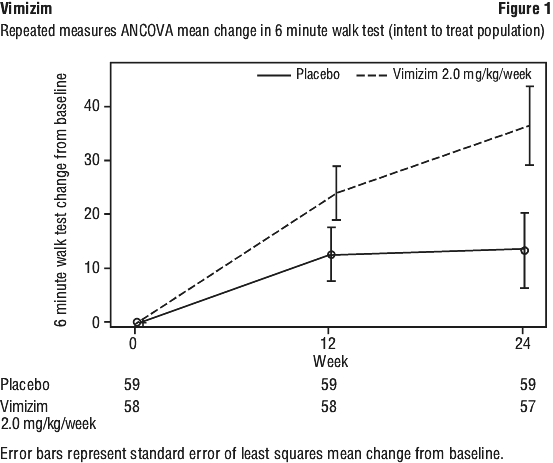
The primary and secondary endpoints were evaluated at Week 24, using an ANCOVA model with treatment, age stratification (5-11, 12-18, ≥ 19 years), and baseline 6-MWT stratification (≤ 200 metres and > 200 metres) as factors. In the intent-to-treat population, the modelled treatment effect in the distance walked in 6 minutes, compared to placebo, was 22.5 m (CI95, 4.0, 40.9; p=0.0174) for the 2 mg/kg/wk regimen. There was no difference in the rate of stair climbing between patients who received Vimizim 2 mg/kg once per week and those who received placebo. Patients who received Vimizim 2 mg/kg once every other week performed similarly in the 6-MWT and 3-MSCT as those who received placebo. The reduction in urinary KS levels from baseline, a measure of pharmacodynamic effect, was greater in the Vimizim treatment groups compared to placebo. The relationship between urinary KS and other measures of clinical response has not been established. The difference was greatest between the placebo group and the weekly treatment group for all endpoints (see Table 3 and Figure 1).
Paediatric population. As for all lysosomal genetic disorders, it is important to initiate treatment as early as possible, before appearance of non-reversible clinical manifestations of the disease.
In an open-label trial (MOR-007), 15 paediatric patients with MPS IVA under the age of 5 years received Vimizim 2 mg/kg every week for 52 weeks. Patients in this study showed a reduction in urinary KS.
5.2 Pharmacokinetic Properties
The pharmacokinetic parameters of Vimizim were evaluated in 23 patients with MPS IVA ranging in age from 5 to 42 years, who received weekly intravenous infusions of 2 mg/kg of Vimizim over approximately 4 hours for 22 weeks and the parameters at Week 0 and Week 22 were compared (see Table 4). At Week 22, the mean AUC0-t and Cmax increased by 181% and 192%, respectively, when compared to Week 0.
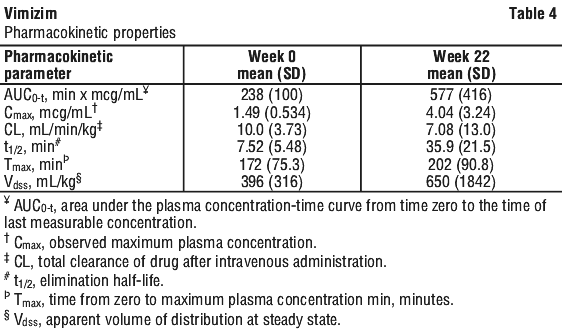
Elimination. Mean half-life (t1/2) increased from 7.52 minutes at week 0 to 35.9 minutes at week 22. Male and female patients had comparable Vimizim clearance, and clearance did not trend with age or weight at Week 22.
The impact of antibodies on Vimizim pharmacokinetics was assessed. No association was apparent between the total antibody titre and Vimizim clearance. However, patients with positive neutralising antibodies responses had decreased total clearance (CL) values and prolonged t1/2. Despite the alteration of the pharmacokinetics profile, presence of neutralising antibodies did not affect pharmacodynamics, efficacy, or safety of the patients who were treated with Vimizim. No accumulation of Vimizim in plasma was evident following weekly dosing.
Metabolism/biotransformation. Elosulfase alfa is a protein and is expected to be metabolically degraded through peptide hydrolysis. Consequently, impaired liver function is not expected to affect the pharmacokinetics of elosulfase alfa.
Excretion. Renal elimination of elosulfase alfa is considered a minor pathway for clearance.
5.3 Preclinical Safety Data
Genotoxicity. Studies to evaluate mutagenic potential have not been performed with Vimizim.
Carcinogenicity. Long-term studies in animals to evaluate carcinogenic potential have not been performed with Vimizim.
6 Pharmaceutical Particulars
6.1 List of Excipients
Sodium acetate trihydrate, monobasic sodium phosphate monohydrate, arginine, hydrochloride, sorbitol, polysorbate 20, water for injections.
6.2 Incompatibilities
This medicine must not be mixed with other medicines except those mentioned in Section 4.2.
6.3 Shelf Life
3 years.
6.4 Special Precautions for Storage
Store at 2°C to 8°C. (Refrigerate. Do not freeze.) Keep the vial in its carton in order to protect from light.
Diluted solutions. Chemical and physical in-use stability has been demonstrated for up to 24 hours refrigerated (2°C - 8°C) followed by up to 24 hours at room temperature (23°C - 27°C). From a microbiological safety point of view, Vimizim should be used immediately. To reduce microbiological hazard, use as soon as possible after dilution. If storage is necessary, hold at 2-8°C for not more than 24 hours.
6.5 Nature and Contents of Container
Clear Type I glass vial with a butyl rubber stopper and an aluminium flip-off crimp seal with a plastic cap. Pack-size of 1 vial.
6.6 Special Precautions for Disposal
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
Chemical structure.

7 Medicine Schedule (Poisons Standard)
S4.
Date of First Approval
09 December 2014
Date of Revision
18 December 2018
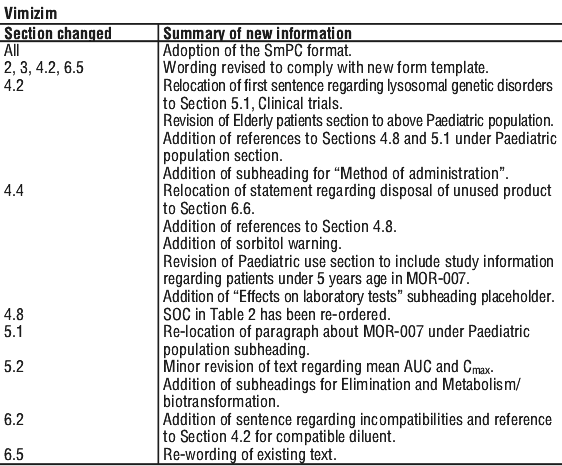
Summary Table of Changes
Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.