Vitrakvi
Brand Information
| Brand name | Vitrakvi |
| Active ingredient | Larotrectinib |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Vitrakvi.
Summary CMI
VITRAKVI®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
▼ This medicine is new or being used differently. Please report side effects. See the full CMI for further details.
1. Why am I using VITRAKVI?
VITRAKVI contains the active ingredient larotrectinib. Vitrakvi (larotrectinib) has provisional approval in Australia for the treatment of adult and paediatric patients from 1 month old, with locally advanced or metastatic solid tumours that:
- have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion without a known acquired resistance mutation,
- are metastatic or where surgical resection is likely to result in severe morbidity, and
- have either progressed following treatment or who have no satisfactory alternative therapy.
These cancers can appear in many different parts of the body.
For more information, see Section 1. Why am I using VITRAKVI? in the full CMI.
2. What should I know before I use VITRAKVI?
Do not use if you have ever had an allergic reaction to VITRAKVI or any of the ingredients listed at the end of the CMI.
Before starting the therapy, a test will be used to determine whether you have NRTK gene alteration.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use VITRAKVI? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with VITRAKVI and affect how it works. Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines including any that you get without a prescription from your pharmacy, supermarket, naturopath or health food shop such as vitamins, dietary supplements or herbal medicine.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use VITRAKVI?
- The recommended dose for adults (from 18 years) is 100 mg, two times a day (= 200mg total per day)
- For babies and children from 1 month old up to 18 years, your child's doctor will work out the right dose for your child.
More instructions can be found in Section 4. How do I use VITRAKVI? in the full CMI.
5. What should I know while using VITRAKVI?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using VITRAKVI? in the full CMI.
6. Are there any side effects?
Like all medicines, VITRAKVI can cause side effects, although not everybody gets them. Tell your doctor or pharmacist if you notice any of the following: feeling tired (fatigue), dizziness, nausea, vomiting, constipation, diarrhea, difficulty in walking, abnormal sense of touch, tingling, change in how things taste, weight increase. For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
▼ This medicine is subject to additional monitoring. This will allow quick identification of new safety information. You can help by reporting any side effects you may get. You can report side effects to your doctor, or directly at www.tga.gov.au/reporting-problems.
VITRAKVI®
Active ingredient(s): larotrectinib
Consumer Medicine Information (CMI)
This leaflet provides important information about using VITRAKVI. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using VITRAKVI.
Where to find information in this leaflet:
1. Why am I using VITRAKVI?
2. What should I know before I use VITRAKVI?
3. What if I am taking other medicines?
4. How do I use VITRAKVI?
5. What should I know while using VITRAKVI?
6. Are there any side effects?
7. Product details
1. Why am I using VITRAKVI?
VITRAKVI contains the active ingredient larotrectinib. Larotrectinib is a highly selective, tropomyosin receptor kinase (TRK) inhibitor and is used to treat a type of cancer called “Tyrosine Receptor Kinase (TRK) fusion cancer”. It works by stopping the TRK fusion proteins working and may slow or stop the cancer growing. It may also help to shrink the cancer.
VITRAKVI has provisional approval in Australia for the treatment of adult and paediatric patients with locally advanced or metastatic solid tumours that:
- have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion without a known acquired resistance mutation,
- are metastatic or where surgical resection is likely to result in severe morbidity, and
- have either progressed following treatment or who have no satisfactory alternative therapy.
2. What should I know before I use VITRAKVI?
Warnings
Do not use VITRAKVI if:
- you are allergic to larotrectinib, or any of the ingredients listed at the end of this leaflet.
Always check the ingredients to make sure you can use this medicine.
Check with your doctor if you:
- have any other medical conditions
- take any medicines for any other condition
- are pregnant or intend to become pregnant.
- breastfeeding or intend to breastfeed
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Laboratory tests
VITRAKVI can increase the amount of some substances in the blood which are made by the liver. Your doctor will do blood tests before and during treatment to check the level of these substances and check how well your liver is working.
Pregnancy and breastfeeding
- You should not use VITRAKVI during pregnancy since the effect of VITRAKVI on the unborn is not known.
- Do not breast-feed while taking this medicine and for 3 days after the last dose. There are no data on the presence of larotrectinib in human milk, the effects of larotrectinib on the breastfed child, or the effects of larotrectinib on milk production.
Contraception – for men and women
- You should avoid getting pregnant while taking this medicine.
- If you are able to become pregnant, your doctor should do a pregnancy test before you start treatment.
- You should also use effective methods of contraception while taking VITRAKVI and for at least one month after the last dose of VITRAKVI. Ask your doctor about the best method of contraception for you.
Children and adolescents
- There is not enough information to recommend the use of this medicine for children under the age of 1 month.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interfere with VITRAKVI and affect how it works.
Medicines that may increase the amount of larotrectinib in your blood and increase the risk of side effect include:
- itraconazole, ketoconazole, posaconazole, fluconazole and voriconazole, clarithromycin, telithromycin - used to treat fungal and bacterial infections
- atazanavir, indinavir, nelfinavir, ritonavir, saquinavir - used to treat HIV infection
- nefadozone - used to treat depression
- diltiazem - used to treat heart conditions and high blood pressure
Medicines that may reduce the amount of larotrectinib in your blood and make VITRAKVI less effective include:
- phenytoin, carbamazepine and phenobarbital - medicines used to treat seizures
- St. John's wort - herbal medicine used to treat depression
- Rifampicin - used to treat bacterial infections
- Efavirenz - used to treat HIV infection
VITRAKVI may affect how some medicines work. Medicines that larotrectinib may decrease the amount of how much of them is found in the blood and make them less effective includes:
- alfentanil - used as a narcotic pain medication
- cyclosporine, sirolimus, tacrolimus - used to prevent organ rejection in patients after transplantation
- Quinidine - used to treat abnormal heart rhythms
- dihydroergotamine, ergotamine - used in migraine or cluster headache attack
- fentanyl - used to treat chronic pain
- pimozide - antipsychotic drug used to control motor or verbal tics
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect VITRAKVI.
4. How do I take VITRAKVI?
How much to take
Adults (from 18 years):
- The recommended dose of VITRAKVI in adults is 100 mg, two times a day (=200 mg total per day).
- You will either take 1 capsule of 100 mg or 4 capsules of 25 mg to make a total dose of 100 mg, two times a day.
- Your doctor will review your dose and change it as needed.
- Follow the instructions provided and use VITRAKVI until your doctor tells you to stop.
Babies and children (from 1 month old up to 18 years):
- Your child's doctor will work out the right dose for your child.
- The maximum recommended dose is 100 mg (maximum of 1 capsule of 100 mg per dose, or 4 capsules of 25 mg per dose, or 5mL of the 20mg/mL solution), two times a day (=200 mg total per day).
- Your child's doctor will review your dose and change it as needed.
- Follow the instructions provided and use VITRAKVI until your child's doctor tells you to stop.
How to take VITRAKVI
- Swallow VITRAKVI capsules whole with a full glass of water.
- Do not open, chew or crush the capsule.
- VITRAKVI can be taken with or without food.
- Do not eat grapefruit or drink grapefruit juice while taking VITRAKVI. This may increase the amount of VITRAKVI in your body.
If you forget to take VITRAKVI
VITRAKVI should be taken regularly at the same time each day. If you miss your dose at the usual time, take your missed dose as soon as you remember prior to the next scheduled dose.
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Do not take a double dose to make up for the dose you missed.
If you take too much VITRAKVI
If you think that you have taken too much VITRAKVI, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while taking VITRAKVI?
Things you should do
Call your doctor straight away if you:
- get any of these symptoms during treatment: feeling dizzy, difficulty walking normally, tingling, feeling numb, or a burning feeling in your hands and feet
- become pregnant during treatment with VITRAKVI or in the first month after your last dose.
Remind any doctor, dentist or pharmacist you visit that you are using VITRAKVI.
Things you should not do
- Do not give this medicine to infants under 1 month of age
- Do not stop using this medicine suddenly or lower the dosage without checking with you doctor.
VITRAKVI with food and drink
Do not eat grapefruit or drink grapefruit juice while taking VITRAKVI. This may increase the amount of VITRAKVI in your body.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how VITRAKVI affects you.
VITRAKVI may cause dizziness, tiredness and nausea in some people. If you have any of these symptoms, do not drive, cycle, use any tools or machines or do anything else that could be dangerous. Children should be careful when riding bicycles or climbing trees.
Drinking alcohol
Tell your doctor if you drink alcohol.
There is no information on the use of VITRAKVI with alcohol.
Looking after your medicine
- For capsules, store below 25°C (77°F).
- For solution, keep refrigerated at 2°C to 8°C (36°F to 46°F). Do not freeze. Use within 30 days of opening.
Follow the instructions in the carton on how to take care of your medicine properly.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Keep it where young children cannot reach it.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Side effects
| Side effects | What to do |
| Call your doctor right away if you notice any of these side effects during treatment |
| Speak to your doctor if you have any of these side effects and they worry you. |
| Common side effects | What to do |
| Speak to your doctor if you have any of these side effects and they worry you. |
Some of these side effects can only be found when your doctor does tests from time to time to check your progress.
| These test results include: | What to do |
| Your doctor will tell you if there are any changes in your blood test that might need treatment. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What VITRAKVI contains
For Capsules:
| Active ingredient | larotrectinib |
| Other ingredients |
|
For Solution:
| Active ingredient | larotrectinib |
| Other ingredients |
|
Do not take this medicine if you are allergic to any of these ingredients.
What VITRAKVI looks like
- VITRAKVI 25 mg hard capsules are white opaque hard gelatin capsule, size 2, with blue printing of “BAYER” cross and “25 mg” on body of capsule (AUST R 320237)
- VITRAKVI 100 mg hard capsules are white opaque hard gelatin capsule, size 0, with blue printing of “BAYER” cross and “100 mg” on body of capsule (AUST R 320238)
- VITRAKVI 20 mg/mL oral solution is a colourless to yellow or orange or red or brownish liquid solution contained in 2 child-resistant glass bottles containing 50 mL oral solution each (AUST R 388084)
Who distributes VITRAKVI
Bayer Australia Limited
ABN 22 000 138 714
875 Pacific Highway Pymble NSW 2073
www.bayer.com.au
This leaflet was prepared on 12 November 2024.
Brand Information
| Brand name | Vitrakvi |
| Active ingredient | Larotrectinib |
| Schedule | S4 |
▼ This medicinal product is subject to additional monitoring in Australia. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse events at www.tga.gov.au/reporting-problems.
MIMS Revision Date: 01 April 2026
1 Name of Medicine
Vitrakvi (larotrectinib).
2 Qualitative and Quantitative Composition
Vitrakvi 25 mg hard capsules. Each capsule contains larotrectinib sulfate, equivalent to 25 mg of larotrectinib.
Vitrakvi 100 mg hard capsules. Each capsule contains larotrectinib sulfate, equivalent to 100 mg of larotrectinib.
Vitrakvi 20 mg/mL oral solution. Each mL oral solution contains larotrectinib sulfate, equivalent to 20 mg of larotrectinib.
For a full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Vitrakvi 25 mg hard capsules. White opaque hard gelatin capsule, size 2, with blue printing of "BAYER" cross and "25 mg" on body of capsule.
Vitrakvi 100 mg hard capsules. White opaque hard gelatin capsule, size 0, with blue printing of "BAYER" cross and "100 mg" on body of capsule.
Vitrakvi 20 mg/mL oral solution. Colourless to yellow or orange or red or brownish liquid solution.
4 Clinical Particulars
4.1 Therapeutic Indications
Vitrakvi (larotrectinib) has provisional approval in Australia for the treatment of adult and paediatric patients with locally advanced or metastatic solid tumours that:
have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion without a known acquired resistance mutation,
are metastatic or where surgical resection is likely to result in severe morbidity, and
have either progressed following treatment or who have no satisfactory alternative therapy.
The decision to approve this indication has been made on the basis of objective response rate (ORR) and duration of response from single-arm clinical studies. The sponsor is required to submit further clinical data to confirm the clinical benefit of the medicine.
4.2 Dose and Method of Administration
Treatment with Vitrakvi should be initiated by physicians experienced in the administration of anticancer therapies.
The presence of an NTRK gene fusion in a tumour specimen should be confirmed by a validated test prior to initiation of treatment with Vitrakvi. (See Section 5.1 Pharmacodynamic Properties, Clinical trials).
Method of administration. For oral use.
Vitrakvi is available as a capsule or oral solution formulation with equivalent oral bioavailability, and may be used interchangeably.
Capsule. The patient should be advised to swallow the capsule whole with water.
Due to the bitter taste of the active ingredient, the capsule should not be opened, chewed, or crushed.
Oral solution. Administer the oral solution by mouth or enterally by naso- or gastric-feeding tube with a dosing syringe.
The oral solution can be administered with or without food using an oral syringe of 1 mL or 5 mL volume or enterally by using a nasogastric feeding tube.
For doses below 1 mL, a 1 mL oral syringe should be used. The calculated dose volume should be rounded to the nearest 0.1 mL.
For doses of 1 mL and higher, a 5 mL oral syringe should be used. The dose volume should be calculated to the nearest 0.2 mL.
Vitrakvi should not be mixed with feeding formulas, if administered via nasogastric feeding tube. Mixing with the feeding formulas could lead to tube blockages.
Instructions for use. Oral syringe. Use a suitable oral syringe with CE marking and bottle adapter (28 mm diameter) if applicable.
For volumes less than 1 mL, use a 1 mL oral syringe with 0.1 mL graduation.
For volumes of 1 mL and higher, use a 5 mL oral syringe with 0.2 mL graduation.
Open the bottle: press the bottle cap and turn it counter-clockwise.
Insert the bottle adapter into the bottle neck and ensure it is well fixed.
Take the oral syringe and ensure that the plunger is fully depressed. Put the oral syringe in the adapter opening. Turn the bottle upside down.
Fill the oral syringe with small amount of solution by pulling the plunger down, then push the plunger upwards to remove any bubbles.
Pull the plunger down to the graduation mark equal to the quantity in mL as prescribed.
Turn the bottle the right way up and remove the oral syringe from the bottle adapter.
Slowly depress the plunger, directing the liquid towards the inside cheek to allow for natural swallowing.
Close the bottle with the original bottle cap (leaving the adapter in place).
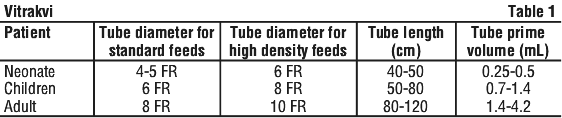
Nasogastric feeding tube. Use a suitable nasogastric feeding tube. The outer diameter of the nasogastric feeding tube should be selected based on the patient characteristics. Typical tube diameter, tube lengths and derived prime volumes are presented in Table 1.
The feeding should be stopped, and the tube flushed with at least 10 mL water.
Note: See exceptions regarding neonates and patients with fluid restrictions in the sub-point directly below.
A suitable syringe should be used to administer Vitrakvi to the nasogastric feeding tube.
The tube should be flushed again with at least 10 mL water to ensure Vitrakvi is delivered and to clear the tube.
Neonates and children with fluid restrictions may require minimal flushing volume of 0.5 to 1 mL or flushing with air to deliver Vitrakvi.
Restart the feeding.
Paediatric. Dosing in paediatric patients is based on body surface area (BSA). The recommended dose of Vitrakvi in paediatric patients (1 month to 18 years) is 100 mg/m2 taken orally, twice daily with a maximum of 100 mg per dose until the patient is no longer clinically benefiting from therapy or until unacceptable toxicity occurs.
Missed dose. If a dose is missed, the patient should not take two doses at the same time to make up for a missed dose. Patients should take the next dose at the next scheduled time. If the patient vomits after taking a dose, the patient should not take an additional dose to make up for vomiting.
Dose modification. Recommended dose modifications for Vitrakvi are provided in Table 2.

For all Grade 2 adverse reactions, continued dosing may be appropriate, though close monitoring to ensure no worsening of the toxicity is advised.
For adverse reactions ≥ Grade 3 not referring to liver function test abnormalities:
Vitrakvi should be withheld until the adverse reaction resolves or improves to baseline or Grade 1. Resume at the next dose modification if resolution occurs within 4 weeks.
Vitrakvi should be permanently discontinued if an adverse reaction does not resolve within 4 weeks.
For dose modifications in case of liver function test abnormalities while receiving therapy with Vitrakvi, see Table 3 (for further information and frequency of laboratory evaluations in patients, see Section 4.4 Special Warnings and Precautions for Use, Hepatotoxicity).

Co-administration with strong or moderate CYP3A4 inducers. Avoid co-administration of strong or moderate CYP3A4 inducers with Vitrakvi. If co-administration of a strong CYP3A4 inducer cannot be avoided, double the Vitrakvi dose. After the inducer has been discontinued for 3 to 5 elimination half-lives, resume the Vitrakvi dose taken prior to initiating the CYP3A4 inducer; see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions.
Patients with hepatic impairment. The starting dose of Vitrakvi should be reduced by 50% in patients with moderate (Child Pugh B) to severe (Child Pugh C) hepatic impairment. No dose adjustment is recommended for patients with mild hepatic impairment (Child Pugh A) (see Section 5.2 Pharmacokinetic Properties).
Patients with renal impairment. Clinical data from a pharmacokinetic study indicate that larotrectinib exposure was increased 1.46-fold in patients with end-stage renal disease (see Section 5.2 Pharmacokinetic Properties). No dose adjustment is required for patients with renal impairment.
Paediatric patients. Clinical data indicate that systemic exposure in paediatric patients (1 month to 18 years of age) at body surface area (BSA) adjusted dose was similar to that in adults given the dose of 100 mg BID (see Section 5.2 Pharmacokinetic Properties).
Elderly. Clinical data indicate that age has no effect on the systemic exposure of larotrectinib (see Section 5.2 Pharmacokinetic Properties). No dose adjustment is necessary in elderly patients.
Gender. Population pharmacokinetic analyses indicate that gender has no effect on the systemic exposure of larotrectinib (see Section 5.2 Pharmacokinetic Properties).
No dose adjustment is necessary.
Body weight. Population pharmacokinetic analyses indicate that body weight has no effect on the systemic exposure of larotrectinib (see Section 5.2 Pharmacokinetic Properties).
No dose adjustment is necessary.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1.
4.4 Special Warnings and Precautions for Use
Neurologic reactions. Neurologic reactions including dizziness, gait disturbance and paraesthesia were reported in patients receiving larotrectinib (see Section 4.8 Adverse Effects (Undesirable Effects)). For the majority of neurologic reactions, onset occurred within the first three months of treatment.
Caution patients about driving and using machines, until they are reasonably certain Vitrakvi therapy does not affect them adversely. Withholding, reducing, or discontinuing Vitrakvi dosing should be considered, depending on the severity and persistence of these symptoms (see Section 4.2 Dose and Method of Administration; Section 4.7 Effects on Ability to Drive and Use Machines).
Hepatotoxicity. Abnormalities of liver function tests including increased ALT, AST, ALP and bilirubin have been observed in patients treated with Vitrakvi (see Section 4.8 Adverse Effects (Undesirable Effects)). The majority of ALT and AST increases occurred in the first 3 months of treatment. Cases of hepatotoxicity with increases in ALT and/or AST of Grade 2, 3 or Grade 4 severity and increases in bilirubin ≥ 2 x ULN have been reported.
In patients with hepatic transaminase elevations, withhold, modify dose or permanently discontinue Vitrakvi based on the severity (see Section 4.2 Dose and Method of Administration).
Monitor for liver function including ALT, AST, ALP and bilirubin, before the first dose, and then every 2 weeks during the first month, then monthly for the next 6 months of treatment, then periodically during treatment. In patients who develop transaminase elevations, more frequent testing as clinically indicated is needed (see Section 4.2 Dose and Method of Administration).
Use in hepatic impairment. See Section 4.2 Dose and Method of Administration, Additional information on special populations, Patients with hepatic impairment.
Use in renal impairment. See Section 4.2 Dose and Method of Administration, Additional information on special populations, Patients with renal impairment.
Use in the elderly. See Section 4.8 Adverse Effects (Undesirable Effects).
Paediatric use. See Section 4.2 Dose and Method of Administration, Additional information on special populations, Paediatric patients.
Effects on laboratory tests. See Section 4.8 Adverse Effects (Undesirable Effects).
4.5 Interactions with Other Medicines and Other Forms of Interactions
Effects of other medicinal products on larotrectinib. Larotrectinib is a substrate of cytochrome P450 (CYP) 3A, P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). Co-administration of Vitrakvi with strong or moderate CYP3A inhibitors, P-gp and BCRP inhibitors (e.g. atazanavir, clarithromycin, indinavir, itraconazole, ketoconazole, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, troleandomycin, voriconazole, grapefruit or grapefruit juice) may increase larotrectinib plasma concentrations.
Co-administration of Vitrakvi with strong or moderate CYP3A inducers and strong P-gp inducers (e.g. carbamazepine, phenobarbital, phenytoin, rifabutin, rifampin, or St. John's wort) may decrease larotrectinib plasma concentrations.
Effect of CYP3A, P-gp and BCRP inhibitors on larotrectinib. Clinical data in healthy adult subjects indicate that co-administration of a single 100 mg Vitrakvi dose with itraconazole (a strong CYP3A inhibitor and P-gp and BCRP inhibitor) 200 mg once daily for 7 days increased larotrectinib Cmax and AUC by 2.8-fold and 4.3-fold, respectively. See Section 4.2 Dose and Method of Administration.
Physiologically based pharmacokinetic (PBPK) modeling predicts co-administration of Vitrakvi with moderate CYP3A4 inhibitors (fluconazole or diltiazem) will increase Vitrakvi Cmax by up to 1.9-fold and AUC by up to 2.7-fold.
Clinical data in healthy adult subjects indicate that co-administration of a single 100 mg Vitrakvi dose with a single dose of 600 mg rifampin (a P-gp and BCRP inhibitor) increased larotrectinib Cmax and AUC by 1.8-fold and 1.7-fold, respectively (see Section 4.2 Dose and Method of Administration).
Effect of CYP3A and P-gp inducer on larotrectinib. Clinical data in healthy adult subjects indicate that co-administration of a single 100 mg dose of Vitrakvi capsules with rifampin (a strong CYP3A and P-gp inducer) 600 mg once daily for 11 days decreased the AUC of larotrectinib by 81% and of Cmax by 71% as compared to Vitrakvi administered alone.
Physiologically based pharmacokinetic (PBPK) modeling predicts co-administration of Vitrakvi with a moderate CYP3A4 inducer (efavirenz) will decrease Cmax of Vitrakvi by 60% and AUC by 72% compared to Vitrakvi administered alone (see Section 4.2 Dose and Method of Administration).
Effect of other transporter inhibitors on larotrectinib. In vitro, larotrectinib is not a substrate for the transporters OAT1, OAT3, OCT1, OCT2, OATP1B1, or OATP1B3.
Effect of gastric pH-elevating agents on larotrectinib. Larotrectinib has pH-dependent solubility. In vitro studies show that in liquid volumes relevant to the gastrointestinal tract (GI) larotrectinib is fully soluble over entire pH range of the GI tract. Therefore, larotrectinib, is unlikely to be affected by pH-modifying agents.
Effect of larotrectinib on CYP3A substrates. Clinical data in healthy adult subjects indicate that co-administration of Vitrakvi (100 mg twice daily for 10 days) increased the Cmax and AUC of oral midazolam (a sensitive CYP3A substrate) 1.7-fold compared to midazolam alone, suggesting that larotrectinib is a weak inhibitor of CYP3A.
Exercise caution with concomitant use of CYP3A substrates with narrow therapeutic range (e.g. alfentanil, cyclosporin, dihydroergotamine, ergotamine, fentanyl, pimozide, quinidine, sirolimus, or tacrolimus) in patients taking Vitrakvi. If concomitant use of these CYP3A substrates with narrow therapeutic range is required in patients taking Vitrakvi, dose modification of the CYP3A substrates may be required due to adverse reactions.
Effect of larotrectinib on other CYP and transporter substrates. In vitro studies indicate that larotrectinib does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6 at clinically relevant concentrations and is unlikely to affect clearance of substrates of these CYPs.
In vitro studies indicate that larotrectinib induces CYP2B6 but does not induce CYP1A2.
In vitro studies indicate that larotrectinib does not inhibit the transporters BCRP, P-gp, OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, BSEP, MATE1 and MATE2-K at clinically relevant concentrations and is unlikely to affect clearance of substrates of these transporters.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. There are no clinical data on the effect of larotrectinib on fertility. No relevant effects on fertility were observed in repeat-dose toxicity studies in rats and monkeys. (See Section 5.3 Preclinical Safety Data).
Use in pregnancy. (Category C)
There are no clinical data on the use of larotrectinib in pregnant women. Administration of larotrectinib to pregnant rats and rabbits during the period of organogenesis demonstrated no teratogenicity or adverse effects on foetal development at doses that produced maternal exposures up to approximately 32 (rats) and 16 (rabbits) times the human exposure (AUC0-24), respectively, at the recommended dose (see Section 5.3 Preclinical Safety Data). Vitrakvi crosses the placenta in animals.
Based on its mechanism of action, the risk of foetal harm cannot be excluded when larotrectinib is administered to a pregnant woman (see Section 5.1 Pharmacodynamic Properties). Advise pregnant women of the potential risk to a foetus.
Childbearing potential/ contraception. Based on the mechanism of action, foetal harm cannot be excluded when administering larotrectinib to a pregnant woman. Females of childbearing potential should have a pregnancy test prior to starting treatment with Vitrakvi.
Advise female patients of reproductive potential to use highly effective contraception during treatment with Vitrakvi and for at least one month after the final dose.
For males of reproductive potential with a non-pregnant female partner of child-bearing potential, advise use of highly effective contraception during treatment with Vitrakvi and for at least one month after the final dose.
Use in lactation. There are no data on the presence of larotrectinib in human milk, the effects of larotrectinib on the breastfed child, or the effects of larotrectinib on milk production. Because of the unknown risk of larotrectinib in nursing infants, advise a nursing woman to discontinue breastfeeding during treatment with larotrectinib and for 3 days (6 plasma half-lives of larotrectinib and its metabolites) following the final dose.
4.7 Effects on Ability to Drive and Use Machines
Neurologic reactions have been reported in patients receiving larotrectinib which may influence the ability to drive and use machines. Caution patients about driving and using machines, until they are reasonably certain Vitrakvi therapy does not affect them adversely (see Section 4.4 Special Warnings and Precautions for Use).
4.8 Adverse Effects (Undesirable Effects)
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems (Australia).
Summary of the safety profile. The safety of Vitrakvi was evaluated in 444 patients (overall safety population) with locally advanced or metastatic solid tumors, irrespective of NTRK gene fusion status, who received at least one dose of Vitrakvi in one of three clinical trials, Studies 1, 2 ("NAVIGATE"), and 3 ("SCOUT"). The majority (91%) of adult patients (18 years and older) received 100 mg Vitrakvi taken twice daily as their starting dose. Three paediatric dose levels were evaluated with 91% of paediatric patients having received a starting dose of 100 mg/m2 (with a maximum of 100 mg) taken twice daily. Median time on treatment for the overall safety population was 11.15 months (range: 0 to 99.4).
The overall safety population characteristics were comprised of patients with a median age of 44 years (range: 0 to 90) with 35% of patients being paediatric patients.
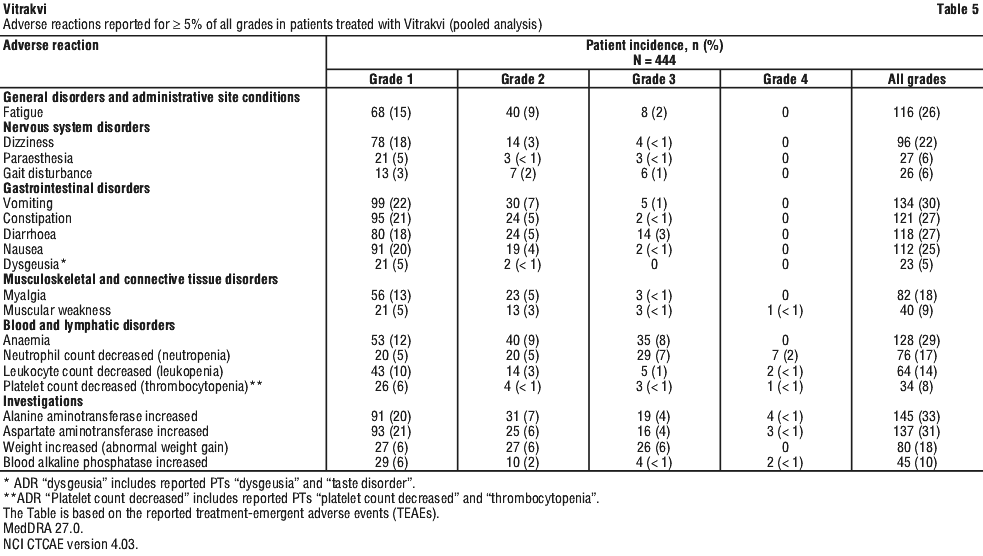
Most commonly (> 20%) reported adverse reactions with Vitrakvi were ALT increased, AST increased, vomiting, anaemia, constipation, diarrhoea, fatigue, nausea, and dizziness.
The majority of adverse reactions were Grade 1 or 2. The highest grades reported with Vitrakvi were Grade 4 neutrophil count decreased, ALT and AST increased, blood alkaline phosphatase increased, leukocyte count decreased, muscular weakness, platelet count decreased and Grade 3 anaemia, weight increased, diarrhoea, fatigue, gait disturbance, vomiting, dizziness, myalgia, paraesthesia, nausea, and constipation. There were no fatal adverse reactions.
Permanent discontinuation of Vitrakvi for treatment emergent adverse events attributed to study drug occurred in 2% of patients (ALT increase, AST increased, amylase increased, emotional poverty, enterocutaneous fistula, gamma-glutamyl transferase increased, hypoventilation, lipase increased, muscular weakness, nausea, and neutrophil count decreased). The majority of events leading to dose reduction occurred in the first three months of treatment.
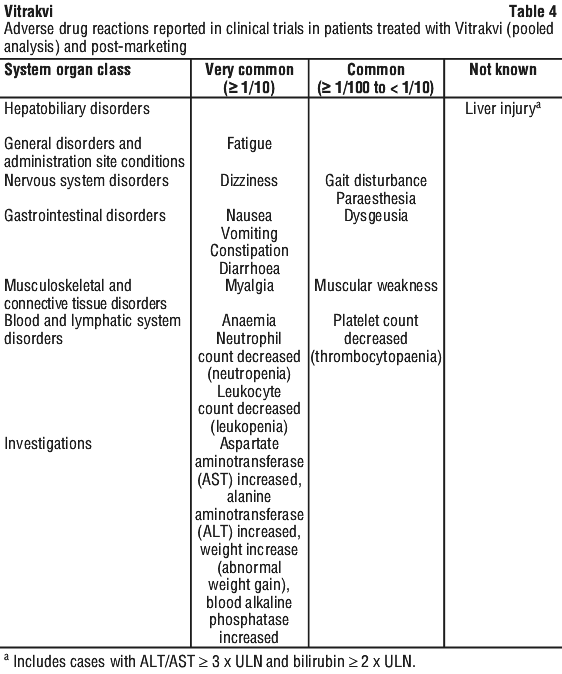
Tabulated list of adverse reactions. The adverse drug reactions reported in the clinical trials and post-marketing in patients treated with Vitrakvi are shown in Table 4. They are classified according to the system organ class (MedDRA version 27.0). The most appropriate MedDRA term is used to describe a certain reaction and its synonyms and related conditions.
Adverse drug reactions are grouped according to their frequencies. Frequency groups are defined by the following convention:
Very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000); not known (cannot be estimated from the available data).
Within each frequency group, undesirable effects are presented in order of decreasing seriousness.
Hepatotoxicity. Abnormalities of liver function tests including increased ALT, AST, ALP and bilirubin have been observed in patients treated with Vitrakvi.
In the overall safety database (N = 444), the maximum grade transaminase elevation observed was Grade 4 ALT increase in four patients (≤ 1%) and AST increase in three patients (< 1%). Grade 3 events occurred in 19 (4%) patients for ALT increased and 16 patients (4%) for AST increased. The majority of Grade 3 and 4 elevations were transient appearing in cycles 1-2 of treatments and resolving to Grade 1 by cycles 3-4. Grade 2 ALT and AST increases were observed in 31 patients (7%) and 25 patients (6%), respectively. Grade 1 ALT and AST increases were observed in 91 (20%) and 93 (21%) patients, respectively. Most Grade 1 and 2 transaminase increases did not worsen through continued non-modified dosing.
ALT and AST increases leading to dose modifications or interruptions occurred in 29 (7%) patients and 26 (6%) patients, respectively (see Section 4.4 Special Warnings and Precautions for Use).
Cases of hepatotoxicity with increases in ALT and/or AST of Grade 2, 3 or Grade 4 severity and increases in bilirubin ≥ 2 x ULN have been reported. In some cases, the dose of Vitrakvi was withheld and restarted at a reduced dose, while in other cases treatment was permanently discontinued (see Section 4.4 Special Warnings and Precautions for Use).
Laboratory test abnormalities. Laboratory relevant adverse reactions which meet the internal ADR criteria are shown in Table 6 for laboratory abnormalities.
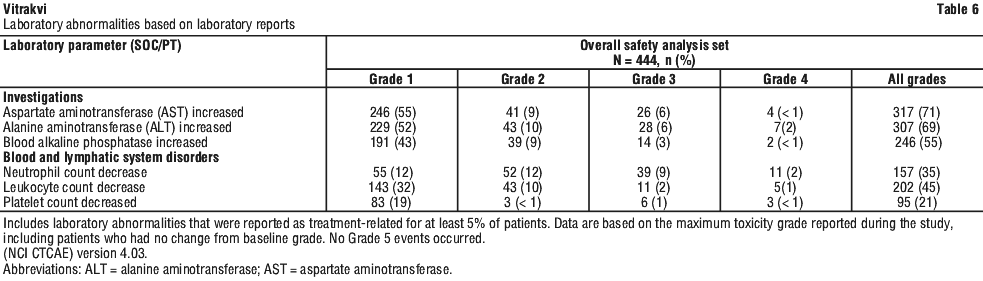
The majority of adverse reactions were Grade 1 or 2 in severity and were resolved without Vitrakvi dose modification or discontinuation. The adverse reactions of vomiting (53% versus 18% in adults), diarrhoea (36% versus 22% in adults), leukocyte count decrease (22% versus 10% in adults), neutrophil count decrease (31% versus 10% in adults), platelet count decreased (12% versus 6% in adults) and transaminase elevations (ALT 42% versus 28% in adults and AST 40% versus 26% in adults) were more frequent in paediatric patients compared to adults. Elevations in liver enzymes in children < 1 years of age may be due to immaturity of liver function.
Elderly patients. Of 444 patients in the overall safety population who received Vitrakvi, 88 (20%) patients were ≥ 65 years of age and 25 (6%) patients were ≥ 75 years of age. Safety profile in elderly patients (≥ 65 years) is consistent with that seen in younger patients (< 65 years). The adverse reactions, dizziness, gait disturbance, fatigue, and anaemia were more frequent in patients ≥ 65 years of age.
4.9 Overdose
For general advice on overdose management, contact the Poisons Information Centre, telephone number 13 11 26 (Australia) or the National Poisons Centre.
There is no known antidote for Vitrakvi. The treatment of overdose with Vitrakvi should consist of general supportive measures.
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Antineoplastic and immunomodulating agents, antineoplastic agents, other antineoplastic agents. ATC code: L01EX12.
Mechanism of action. Larotrectinib is an orally-bioavailable, adenosine triphosphate (ATP)-competitive, potent and highly selective TRK kinase inhibitor that was rationally designed to avoid activity with off-target kinase. The target for larotrectinib is the TRK family of proteins inclusive of TRKA, TRKB, and TRKC that are encoded by NTRK1, NTRK2 and NTRK3 genes, respectively.
In-frame gene fusion events resulting from chromosomal rearrangements of the human genes NTRK1, NTRK2, and NTRK3 lead to the formation of oncogenic TRK fusion proteins. These resultant novel chimeric oncogenic proteins are aberrantly expressed driving constitutive kinase activity subsequently activating downstream cell signaling pathways involved in cell proliferation and survival leading to TRK fusion cancer.
Larotrectinib demonstrated potent inhibition of TRK proteins and inhibition of proliferation of tumor cells in a concentration-dependent manner. In TRK fusion-driven mouse xenograft models larotrectinib treatment induced a significant reduction of tumor growth.
Acquired resistance mutations after progression on TRK inhibitors have been observed. Larotrectinib had minimal activity in cell lines with point mutations in the TRKA kinase domain, including the clinically identified acquired resistance mutation, G595R. Point mutation in the TRKC kinase domain with clinically identified acquired resistance to larotrectinib include G623R, G696A, and F617L.
Pharmacodynamic effects. Cardiac electrophysiology. In 36 healthy adult subjects receiving single doses ranging from 100 mg to 900 mg, Vitrakvi did not prolong the QT interval to any clinically relevant extent and there was no relationship between exposure (Cmax) and changes in the QT interval.
Clinical trials. The efficacy and safety of Vitrakvi were demonstrated in three multicentre, open-label, single-arm clinical studies in adult and paediatric cancer patients (Table 7).
Patients with and without documented NTRK gene fusion were allowed to participate in Study 1 and Study 3 ("SCOUT"). Patients enrolled to Study 2 ("NAVIGATE") were required to have TRK fusion cancer. The pooled analysis set of efficacy includes 164 patients with TRK fusion cancer enrolled across the three studies who had measurable disease assessed by RECIST, a non-CNS primary tumor and received at least one dose of larotrectinib. These patients were required to have received prior standard therapy appropriate for their tumor type and stage of disease or who, in the opinion of the Investigator, would have had to undergo radical surgery (such as limb amputation, facial resection, or paralysis causing procedure), or were unlikely to tolerate, or derive clinically meaningful benefit from available standard of care therapies in the advanced disease setting. Identification of NTRK gene fusions relied upon the molecular test methods next generation sequencing (NGS), reverse transcription-polymerase chain reaction (RT-PCR) and fluorescence in situ hybridization (FISH) as routinely performed at Clinical Laboratory Improvement Amendments (CLIA) or other similarly certified laboratories.
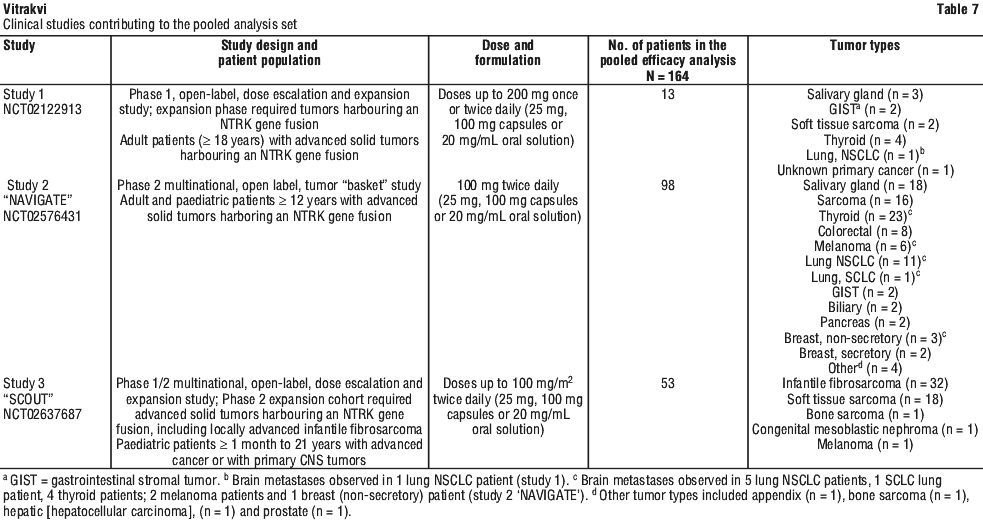
Baseline characteristics for the pooled 164 patients with solid tumors harbouring an NTRK gene fusion were as follows: median age 42 years (range 0.1-84 years); 34% < 18 years of age, and 66% ≥ 18 years; 77% White and 49% male; and ECOG PS* 0-1 (86%), 2 (12%), or 3 (2%). Ninety-four percent of patients had received prior treatment for their cancer, defined as surgery, radiotherapy, or systemic therapy. Of these, 77% had received prior systemic therapy with a median of 2 prior systemic treatment regimens received. Twenty seven percent of all patients had received 3 or more prior systemic therapies and 51% of all patients had received 1-2 prior systemic therapies. Twenty two percent of all patients had received no prior systemic therapy.
The most common tumor types represented were soft tissue sarcoma (22%), infantile fibrosarcoma (20%), thyroid cancer (16%), salivary gland tumor (13%) and lung cancer (8%).
* Eastern Cooperative Oncology Group (ECOG) Performance Status.
Efficacy results. The pooled efficacy results for overall response rate, duration of response and time to first response, stable disease and disease control rate are presented in Table 8.
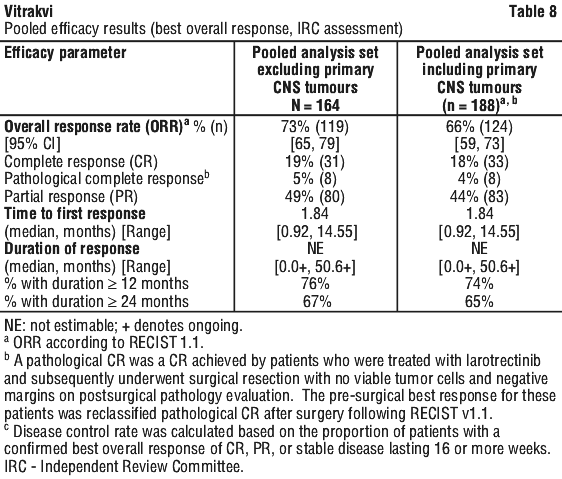
Pooled primary analysis set. At the time of analysis, the median duration of response had not been reached, an estimated 76% [95% CI: 67, 85] of responses lasted 12 months or longer and 67% [95% CI: 55, 78] of responses lasted 24 months or longer. The disease control rate is 84% (137/164) [95% CI: 77, 89]. Ninety percent (90%) [95% CI: 85, 95] of patients treated were alive one year after the start of therapy and 82% [95% CI: 75, 90] after two years with the median for overall survival not yet being reached. Median progression free survival was 33.4 months at the time of the analysis, with a progression free survival rate of 66% [95% CI: 58, 74] after 1 year and 58% [95% CI: 48, 67] after 2 years.
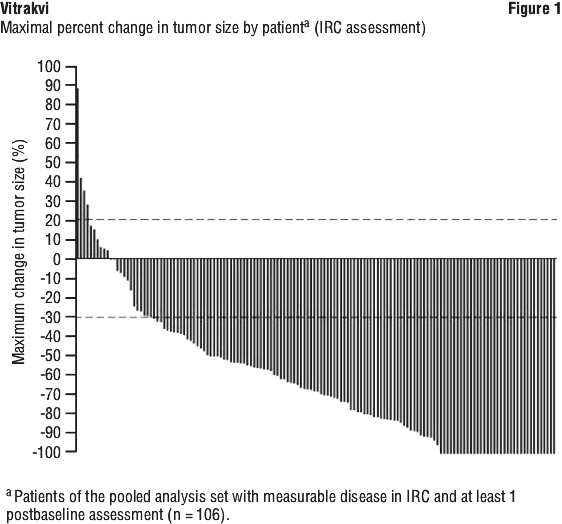
A reduction in tumor size was observed in 81% (n = 144) patients with NTRK gene fusions, regardless of age, prior treatment, tumor type, NTRK gene involved or NTRK gene fusion partner. Changes in target lesion size are illustrated in a Waterfall plot in Figure 1. The median change in tumor size was a decrease of 68%.
Of the 24 patients with primary CNS confirmed response was observed in 5 patients (21%) with 2 of the 24 patients (8%) being complete responders and 3 (12.5%) being partial responders. In 2 additional patients (8%) a not yet confirmed partial response was observed. Further, 15 patients (63%) had stable disease. Two patients (8%) had a progressive disease.
At the time of data cut-off, time on treatment ranged from 1.2 to 21.4 months and was ongoing in 15 out of 24 patients with one of these patients receiving post-progression treatment.
Health-related quality of life outcomes. Paediatric and adult patients with TRK fusion cancer treated with Vitrakvi experienced clinically meaningful improvement in health-related quality of life (HRQoL) as evaluated by the EORTC QLQ-C30, EQ-5D 5L VAS and PedsQL questionnaires. Within 2 months of treatment, rapid and sustained improvements were seen in the majority of the functional domains of the questionnaires. Overall health status results are presented in Table 9.

5.2 Pharmacokinetic Properties
In cancer patients given Vitrakvi capsules, peak plasma levels (Cmax) of larotrectinib were achieved at approximately 1 hour after dosing. Half-life (t1/2) is approximately 3 hours and steady state is reached within 8 days. Systemic accumulation is 1.6-fold at steady state. At the recommended dose of 100 mg taken twice daily, steady-state arithmetic mean (± standard deviation) Cmax and daily AUC in adults were 914 ± 445 nanogram/mL and 5410 ± 3813 nanogram*h/mL, respectively.
Absorption. Vitrakvi is available as a capsule and oral solution formulation.
The mean absolute bioavailability of larotrectinib was 34% (range: 32% to 37%) following a single 100 mg oral dose.
In healthy adult subjects, the bioavailability of larotrectinib in the oral solution formulation was similar to the capsule, with Cmax 36% higher with the oral solution formulation.
Larotrectinib Cmax was reduced by approximately 35% and there was no effect on AUC in healthy subjects administered Vitrakvi after a high-fat and high-calorie meal compared to the Cmax and AUC after overnight fasting.
Distribution. The mean volume of distribution (Vss) of larotrectinib in healthy adult subjects was 48 L following intravenous administration of an IV microtracer in conjunction with a 100 mg oral dose. Binding of larotrectinib to human plasma proteins in vitro was approximately 70% and was independent of drug concentration. The blood-to-plasma concentration ratio was approximately 0.9.
Metabolism. Larotrectinib was metabolized predominantly by CYP3A4/5 in vitro. Following oral administration of a single 100 mg dose of radiolabeled larotrectinib to healthy adult subjects, unchanged larotrectinib (19%) and an O-glucuronide that is formed following loss of the hydroxypyrrolidine-urea moiety (26%) were the major circulating radioactive drug components.
Excretion. The half-life of larotrectinib in plasma of cancer patients given 100 mg twice daily of Vitrakvi was approximately 3 hours. Mean clearance (CL) of larotrectinib was approximately 34 L/h following intravenous administration of an IV microtracer in conjunction with a 100 mg oral dose of Vitrakvi.
Following oral administration of 100 mg radiolabeled larotrectinib to healthy adult subjects, 58% of the administered radioactivity was recovered in faeces and 39% was recovered in urine. The fraction excreted as unchanged drug in urine was 29% following IV microtracer dose, indicating that direct renal excretion accounted for 29% of total clearance.
Additional information on special populations. Paediatric patients. Based on population pharmacokinetic analyses exposure (Cmax and AUC) in paediatric patients (1 month to < 3 months of age) at the recommended dose of 100 mg/m2 with a maximum of 100 mg BID was 3-fold higher than in adults (≥ 18 years of age) given the dose of 100 mg BID. At the recommended dose, the Cmax in paediatric patients (≥ 3 months to < 12 years of age) was higher than in adults, but the AUC was similar to that in adults. For paediatric patients older than 12 years of age, the recommended dose is likely to give similar Cmax and AUC as observed in adults.
Data defining exposure in small children (1 month to < 6 years of age) at the recommended dose is limited (n = 33).
Elderly. AUC in patients > 65 years or > 80 years were similar to those in younger patients (< 65 years).
Patients with hepatic impairment. A pharmacokinetic study was conducted in subjects with mild (Child Pugh A), moderate (Child Pugh B) and severe (Child Pugh C) hepatic impairment, and in healthy adult control subjects with normal hepatic function matched for age, body mass index and sex. All subjects received a single 100 mg dose of larotrectinib. An increase in larotrectinib AUC0-inf was observed in subjects with mild, moderate and severe hepatic impairment of 1.3, 2 and 3.2-fold, respectively, versus those with normal hepatic function. Cmax was observed to increase slightly by 1.1, 1.1 and 1.5-fold, respectively.
Patients with renal impairment. A pharmacokinetic study was conducted in subjects with end stage renal disease requiring dialysis, and in healthy adult control subjects with normal renal function matched for age, body mass index and sex. All subjects received a single 100 mg dose of larotrectinib. An increase in larotrectinib Cmax and AUC0-inf, of 1.25 and 1.46-fold, respectively, was observed in renally impaired subjects versus those with normal renal function.
5.3 Preclinical Safety Data
The safety pharmacology of larotrectinib was evaluated in several in vitro and in vivo studies that assessed effects on the CV, CNS, respiratory, and GI systems in various species (rat, mouse, dog, cynomolgus monkey). Larotrectinib had no adverse effects on hemodynamic parameters and ECG intervals in telemetered monkeys at exposures (Cmax) which are approximately 6-fold the human therapeutic exposures. Larotrectinib had no neurobehavioral findings in rats and did not affect neuromuscular function in mice.
Larotrectinib had no effects on respiratory function in rats; at exposures (Cmax) at least 8-times the human therapeutic exposures. In rats, larotrectinib accelerated intestinal transit and increased gastric secretion and acidity.
Systemic toxicity was assessed in studies with daily oral administration up to 13-weeks in rats and monkeys. Dose limiting skin lesions were only seen in rats and were primarily responsible for mortality and morbidity. Skin lesions were not seen in monkeys.
Clinical signs of gastrointestinal toxicity were dose limiting in monkeys. The following additional relevant findings were observed in both species: increased body weight and food consumption; an adaptive response in the liver secondary to induction of drug metabolizing enzymes; elevated serum liver enzymes (ALT and/or AST); histopathological changes in lymphoid tissues without corresponding changes in white blood cell counts; in rats, pancreatic changes; minor, reversible changes on red blood cell parameters; and increased heart weights without histopathological correlate were reported. In rats severe toxicity (STD10) was observed at doses corresponding to 1- to 2-times the human AUC at the recommended clinical dose. No relevant systemic toxicity was observed in monkeys at exposures which correspond to > 10-times the human AUC at the recommended clinical dose.
Larotrectinib was administered to juvenile rats from postnatal day (PND) 7 to 70. Pre-weaning mortality (before PND 21) was observed at the high dose corresponding to 2.5- to 4-times the AUC at the recommended dose. Growth and nervous system effects were seen at 0.5- to 4-times the AUC at the recommended dose. The male growth reduction was associated with delayed puberty. Nervous system effects and reduced body weight in females demonstrated partial recovery.
Larotrectinib was not teratogenic and embryotoxic when dosed daily during the period of organogenesis to pregnant rats and rabbits at maternotoxic doses, i.e. corresponding to 32-times (rats) and 16-times (rabbits) the human AUC at the recommended clinical dose. Larotrectinib crosses the placenta in both species.
Fertility studies with larotrectinib have not been conducted. Larotrectinib had no effects on spermatogenesis in rats and on male reproductive organs in rats and monkeys at doses corresponding to approximately 2-times (rats) and 10-times (monkeys) the human AUC at the recommended clinical dose.
In a 1-month study in rats, fewer corpora lutea, increased incidence of anestrus and decreased uterine weight with uterine atrophy were observed and these effects were reversible. No effects on female reproductive organs were seen in the 13-week study in rats and monkeys at doses corresponding to approximately 3-times (rats) and approximately 17-times (monkeys) the human AUC at the recommended clinical dose.
In juvenile animals, a decrease in pregnancy rate was reported despite normal mating at the high-dose corresponding to 3-times the AUC at the recommended paediatric dose.
Genotoxicity. Larotrectinib was not mutagenic in bacterial reverse mutation (Ames) assays and in in vitro mammalian mutagenesis assays. Larotrectinib was negative in the in vivo mouse micronucleus test.
Carcinogenicity. Carcinogenicity studies have not been performed with larotrectinib.
6 Pharmaceutical Particulars
6.1 List of Excipients
Capsules. Gelatin; titanium dioxide; printing ink - blue: shellac, FD and C Blue # 2 aluminium lake, titanium dioxide, propylene glycol, ammonia solution, dimethicone.
Oral solution. Purified water, hydroxypropyl betadex, sucralose, sodium citrate, sodium benzoate, strawberry flavour, citric acid.
6.2 Incompatibilities
Not applicable.
6.3 Shelf Life
Information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Capsules. Store below 25°C; temperature excursions between 15°C and 30°C (59°F to 86°F) are permitted.
Oral solution. Store at 2° to 8°C (36° to 46°F). Refrigerate. Do not freeze.
6.5 Nature and Contents of Container
Capsule. 56 counted (HDPE) bottle closed with polypropylene (PP) closure.
Oral solution. Each carton contains two amber glass bottles containing 50 mL oral solution each and capped with a child-resistant polypropylene (PP) screw cap.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
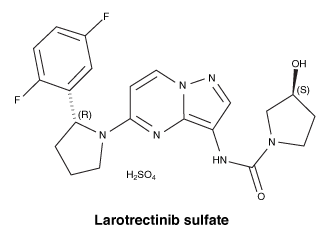
Empirical name: C21H24F2N6O6S (sulfate salt form).
Molecular weight: 526.51 g/mol (sulfate salt form).
7 Medicine Schedule (Poisons Standard)
S4 Prescription Only Medicine.
Date of First Approval
07 September 2020
Date of Revision
03 February 2026
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.