Vyndamax
Brand Information
| Brand name | Vyndamax |
| Active ingredient | Tafamidis |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Vyndamax.
Summary CMI
Vyndamax®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
▼ This medicine is new or being used differently. Please report side effects. See the full CMI for further details.
1. Why am I using Vyndamax?
Vyndamax contains the active ingredient tafamidis. Vyndamax is used to treat a disease called transthyretin amyloid cardiomyopathy, also known as ATTR-CM.
For more information, see Section 1. Why am I using Vyndamax? in the full CMI.
2. What should I know before I use Vyndamax?
Do not use if you have ever had an allergic reaction to Vyndamax or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use Vyndamax? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with Vyndamax and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use Vyndamax?
- Take one 61 mg capsule once a day.
More instructions can be found in Section 4. How do I use Vyndamax? in the full CMI.
5. What should I know while using Vyndamax?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using Vyndamax? in the full CMI.
6. Are there any side effects?
Common side effects include diarrhoea feeling weak or a lack of energy, feeling unbalanced when standing or walking, fall, sinusitis, cataract (clouding of the lens in your eye), flatulence, muscle or joint pain, skin ulcer, bladder infections and excessive sweating.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
▼ This medicine is subject to additional monitoring. This will allow quick identification of new safety information. You can help by reporting any side effects you may get. You can report side effects to your doctor, or directly at www.tga.gov.au/safety/reporting-problems.
Vyndamax® (vin-der-max)
Active ingredient(s): tafamidis (ter-fam-e-dis)
Consumer Medicine Information (CMI)
This leaflet provides important information about using Vyndamax. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using Vyndamax.
Where to find information in this leaflet:
1. Why am I using Vyndamax?
2. What should I know before I use Vyndamax?
3. What if I am taking other medicines?
4. How do I use Vyndamax?
5. What should I know while using Vyndamax?
6. Are there any side effects?
7. Product details
1. Why am I using Vyndamax?
Vyndamax contains the active ingredient tafamidis.
Vyndamax is used to treat a disease called transthyretin amyloid cardiomyopathy, also known as ATTR-CM.
In patients with ATTR-CM, a protein called transthyretin (TTR) breaks up and may form fibrils called amyloid. Amyloid can build up between cells in your heart and in other places in your body, preventing your heart from working normally and causing symptoms.
Vyndamax is used to prevent TTR from breaking up and forming amyloid.
This medicine belongs to a group of medicines called transthyretin stabilisers.
Ask your doctor if you have any questions about why this medicine has been prescribed for you.
Your doctor may have prescribed it for another reason.
2. What should I know before I use Vyndamax?
Do not use Vyndamax if:
- You are allergic to tafamidis, or any of the ingredients listed at the end of this leaflet.
Always check the ingredients to make sure you can use this medicine.
Some of the symptoms of an allergic reaction may include shortness of breath, wheezing or difficulty breathing, swelling of the face, lips, tongue or other parts of the body and/or rash, itching or hives on the skin.
- You are pregnant.
- You are breast feeding.
- You are aged under 18 years.
Check with your doctor if you:
- Have any other medical conditions including:
- An illness requiring an organ transplant
- A medical condition affected by excessive sorbitol
- Severe liver problems
- Severe kidney problems. - Have allergies to any other medicines, foods, preservatives or dyes
- Take any medicines for any other condition.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
If you are able to become pregnant, you must use birth control while you are taking Vyndamax and should continue using birth control for one month after stopping treatment with Vyndamax.
There are no data on the use of Vyndamax in pregnant women.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interfere with Vyndamax and affect how it works. These include:
- Methotrexate - a medicine used to treat rheumatoid arthritis or some cancers
- Rosuvastatin, atorvastatin - medicines used to treat high cholesterol
- Apixaban, rivaroxaban - medicines used to thin blood
- Imatinib - a medicine used for some cancer treatment.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect Vyndamax.
4. How do I use Vyndamax?
How much to take
- Take one 61 mg capsule once a day.
- Follow the instructions provided and use Vyndamax until your doctor tells you to stop.
One 61 mg Vyndamax capsule will produce the same level of active ingredient in your blood and give the same effect as four 20 mg capsules of Vyndaqel (total dose = 80 mg), even though one milligram of Vyndamax is not the same as one milligram of Vyndaqel.
Do not take Vyndamax and Vyndaqel at the same time.
When to take Vyndamax
- Take your medicine at about the same time each day.
- It does not matter if you take this medicine with or without food.
How to take Vyndamax
- Swallow the capsule whole with a full glass of water.
- The capsule should not be crushed or cut.
If you forget to take Vyndamax
Vyndamax should be used regularly at the same time each day. If you miss your dose at the usual time, and if it is within 6 hours before your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take it as soon as you remember, and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for the dose you missed.
If you take too much Vyndamax
If you think that you have taken too much Vyndamax, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using Vyndamax?
Things you should do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Vyndamax.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine.
If you become pregnant while taking this medicine, tell your doctor immediately.
If you are about to have any blood tests, tell your doctor that you are taking this medicine.
Keep all of your doctor's appointments so that your progress can be checked.
Things you should not do
Do not take Vyndamax to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how Vyndamax affects you.
Looking after your medicine
- Keep your capsules in the pack until it is time to take them.
- Keep your capsules in a cool dry place where the temperature stays below 25°C.
Follow the instructions in the carton on how to take care of your medicine properly.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Do not use this medicine after the expiry date.
Keep it where young children cannot reach it.
When to discard your medicine
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Side effects
| Side effects | What to do |
| Speak to your doctor if you have any of these side effects and they worry you. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/safety/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What Vyndamax contains
| Active ingredient (main ingredient) | 61 mg tafamidis |
| Other ingredients (inactive ingredients) | Butylated hydroxytoluene Ethanol Gelatin Glycerol Iron oxide red Isopropyl alcohol Polysorbate 20 Polyvinyl acetate phthalate Povidone Propylene glycol Macrogol 400 Mannitol Sorbitol Strong ammonium solution Titanium dioxide |
Do not take this medicine if you are allergic to any of these ingredients.
This medicine contains sulfites.
This medicine does not contain lactose, sucrose, gluten, tartrazine or any other azo dyes.
What Vyndamax looks like
Vyndamax 61 mg is a reddish brown, opaque, oblong capsule printed with "VYN 61" in white.
They are supplied in blister packs of 30 capsules.
AUST R 314813
Who distributes Vyndamax
Pfizer Australia Pty Ltd
Sydney, NSW
Toll Free Number: 1800 675 229
www.pfizermedicalinformation.com.au
This leaflet was prepared in January 2026.
® Registered Trademark
© Pfizer Australia Pty Ltd 2026
Brand Information
| Brand name | Vyndamax |
| Active ingredient | Tafamidis |
| Schedule | S4 |
▼ This medicinal product is subject to additional monitoring in Australia. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse events at www.tga.gov.au/reporting-problems.
MIMS Revision Date: 01 March 2026
1 Name of Medicine
Tafamidis.
2 Qualitative and Quantitative Composition
The drug product is a soft capsule containing 61 mg micronised tafamidis.
Each soft capsule contains no more than 44 mg of sorbitol.
The soft capsules contain sulfites.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Soft capsule.
Reddish brown, opaque, oblong (approximately 21 mm) capsule printed with "VYN 61" in white.
4 Clinical Particulars
4.1 Therapeutic Indications
Vyndamax is indicated for the treatment of adult patients with wild-type or hereditary transthyretin amyloid cardiomyopathy (ATTR-CM).
4.2 Dose and Method of Administration
Treatment should be initiated and remain under the supervision of a physician knowledgeable in the management of patients with transthyretin amyloid cardiomyopathy (ATTR-CM).
Dosage. The recommended dose of Vyndamax is 61 mg tafamidis orally once daily (see Section 5.1 Pharmacodynamic Properties).
A single 61 mg Vyndamax (tafamidis) capsule is bioequivalent to 80 mg Vyndaqel (tafamidis meglumine) (administered as four 20 mg Vyndaqel capsules) and is not interchangeable on a per mg basis (see Section 5.1 Pharmacodynamic Properties; Section 5.2 Pharmacokinetic Properties).
No dose ranging studies have been undertaken.
Method of administration. Oral use.
The capsule(s) should be swallowed whole and not crushed or cut. Vyndamax may be taken with or without food.
If a dose is missed, the patient should take the dose as soon as remembered. If it is almost time for the next dose, the patient should skip the missed dose and take the next dose at the regularly scheduled time. Do not double the dose.
Dosage adjustment. Renal or hepatic impairment. No dosage adjustment is required for patients with renal impairment, or mild or moderate hepatic impairment. Vyndamax has not been studied in patients with severe hepatic impairment (see Section 5.2 Pharmacokinetic Properties).
Elderly. No dosage adjustment is required for elderly patients (≥ 65 years) (see Section 5.2 Pharmacokinetic Properties). Of the total number of patients in the clinical study (n=441), 90.5% were 65 and over, with a median age of 75 years.
Paediatric. The safety and effectiveness of Vyndamax have not been established in paediatric patients.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients of Vyndamax listed in Section 6.1 List of Excipients.
4.4 Special Warnings and Precautions for Use
Women of childbearing potential. Studies in animals have shown developmental toxicity (see Section 4.6 Fertility, Pregnancy and Lactation). The potential risk for humans is unknown. Vyndamax is not recommended during pregnancy. Women of childbearing potential should use appropriate contraception when taking Vyndamax and continue to use appropriate contraception for 1-month after stopping treatment with Vyndamax (see Section 4.6 Fertility, Pregnancy and Lactation).
Organ transplant patients. A study has not been conducted in organ transplant patients. The efficacy and safety of Vyndamax in organ transplant patients has not been established.
BCRP substrates. Based on the potential of Vyndamax to inhibit the efflux transporter BCRP (breast cancer resistant protein), caution should be used when co-administering Vyndamax and BCRP substrates, because of the risk of BCRP substrate related adverse reactions (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Sorbitol. This medicinal product contains no more than 44 mg sorbitol in each capsule.
The additive effect of concomitantly administered products containing sorbitol (or fructose) and dietary intake of sorbitol (or fructose) should be taken into account.
The content of sorbitol in medicinal products for oral use may affect the bioavailability of other medicinal products for oral use administered concomitantly.
Liver function tests. Increase in liver function tests may occur (see Section 4.8 Adverse Effects (Undesirable Effects)).
Use in hepatic impairment. Tafamidis has not been studied in patients with severe hepatic impairment and caution is recommended (see Section 5.2 Pharmacokinetic Properties).
Use in renal impairment. Limited data are available in patients with severe renal impairment (creatinine clearance less than or equal to 30 mL/min).
Use in the elderly. See Section 4.2 Dose and Method of Administration.
Paediatric use. See Section 4.2 Dose and Method of Administration.
Effects on laboratory tests. Tafamidis may decrease serum concentrations of total thyroxine, without an accompanying change in free thyroxine (T4) or thyroid stimulating hormone (TSH). This observation in total thyroxine values may likely be the result of reduced thyroxine binding to or displacement from transthyretin (TTR) due to the high binding affinity tafamidis has to the TTR thyroxine receptor. No corresponding clinical findings consistent with thyroid dysfunction have been observed (see Section 4.8 Adverse Effects (Undesirable Effects)).
The incidence of thyroxine abnormality < 0.8 x Lower Level of Normal (LLN) was greater in the tafamidis meglumine 80 mg group (29.7%) than in the tafamidis meglumine 20 mg (12.3%) and placebo (4.5%) groups.
4.5 Interactions with Other Medicines and Other Forms of Interactions
In a clinical study in healthy volunteers, tafamidis meglumine did not induce or inhibit the cytochrome P450 enzyme CYP3A4.
In vitro data also indicated that tafamidis does not significantly inhibit cytochrome P450 enzymes CYP1A2, CYP3A4, CYP3A5, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP2D6. In addition, tafamidis did not induce CYP1A2, but did induce CYP2B6 in vitro, however based on the negative clinical CYP3A4 induction results, it can be concluded that the likelihood of CYP2B6 clinical induction is low.
In vitro studies suggest that it is unlikely tafamidis will cause drug interactions at clinically relevant concentrations with substrates of UDP-glucuronosyltransferase (UGT) systemically. Tafamidis may inhibit intestinal activities of UGT1A1.
Tafamidis showed a low potential to inhibit multi-drug resistant protein (MDR1) (also known as P-glycoprotein; P-gp) systemically and in the gastrointestinal (GI) tract, organic cation transporter 2 (OCT2), multidrug and toxin extrusion transporter 1 (MATE1) and MATE2K, organic anion transporting polypeptide 1B1 (OATP1B1) and OATP1B3 at clinically relevant concentrations.
Tafamidis has the potential to inhibit the efflux transporter BCRP and may increase systemic exposure of substrates of this transporter (e.g. methotrexate, rosuvastatin, atorvastatin, apixaban, rivaroxaban, imatinib). In a clinical study in healthy participants, the exposure of the BCRP substrate rosuvastatin increased approximately 2-fold following multiple doses of 61 mg tafamidis daily dosing. Dose adjustment may be needed for these substrates.
Patients should be carefully monitored for BCRP substrate related adverse reactions when used concomitantly with Vyndamax. A dose modification of the BCRP substrate according to its Prescribing Information should be considered.
Tafamidis may have the potential to inhibit organic anion transporter 1 (OAT1) and OAT3 and may cause drug-drug interactions with substrates of these transporters (e.g. non-steroidal anti-inflammatory drugs, bumetanide, furosemide, lamivudine, methotrexate, oseltamivir, tenofovir, ganciclovir, adefovir, cidofovir, zidovudine, zalcitabine). However, additional risk assessments based on the R-value model (AUCi/AUC =1+(Cmax,u/Ki)) were performed and the maximal predicted changes in AUC of OAT1 and OAT3 substrates were determined to be less than 1.25 for the 61 mg tafamidis daily dose, therefore, inhibition of OAT1 or OAT3 transporters by tafamidis is not expected to result in clinically significant interactions.
Patients receiving substrates of both BCRP and OAT with tafamidis should be assessed as exposure of these drugs may be increased e.g. methotrexate AUC might be increased by ~50%.
No interaction studies have been performed evaluating the effect of other medicinal products on tafamidis.
No significant effect was observed on the pharmacokinetics of midazolam (a CYP3A4 substrate) or on the formation of its active metabolite (1-hydroxymidazolam), when a single 7.5 mg dose of midazolam was administered prior to and after a 14-day regimen of 20 mg once-daily tafamidis meglumine. The overall systemic exposure (AUC0-∞) and total clearance (CL/F) of midazolam were shown to be equivalent. In addition, tafamidis did not induce CYP3A4 activity in either male or female subjects.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. There were no effects of tafamidis meglumine on fertility, reproductive performance, or mating behavior in the rat at any dose. Rats were dosed daily (5, 15, and 30 mg/kg/day) prior to cohabitation (for at least 15 days for females and 28 days for males), throughout the cohabitation period to the day prior to termination of males and through to implantation of females (Gestation Day 7). No adverse effects were noted on male rats in toxicity, fertility and mating behaviour at any dose. Because no reproductive effects occurred at the highest dose tested, the paternal and maternal no observed effect level for reproductive toxicity of tafamidis meglumine is greater than 30 mg/kg/day (9.5-times the clinical AUC at the MRHD of 61 mg tafamidis per day).
Developmental toxicity. In pregnant rabbits increased skeletal variations were observed at ≥ 0.5 mg/kg/day (exposures approximately equivalent to clinical exposures at the MRHD of 61 mg tafamidis), while increased skeletal malformations, reduced embryo-fetal survival and reduction in fetal body weights were observed at 8 mg/kg/day (AUC exposures ≥ 9.1-times clinical AUC at the MRHD). In pregnant rats, oral administration of tafamidis (15, 30, and 45 mg/kg/day) from Gestation Day 7 through 17 resulted in decreased fetal weights at ≥ 30 mg/kg/day (approximately ≥ 9.5-times the human AUC at the clinical dose of 61 mg tafamidis).
In the rat pre- and post-natal development study with tafamidis, pregnant rats were orally administered tafamidis meglumine at doses of 5, 15, or 30 mg/kg/day from Gestation Day 7 through Lactation Day 20. Decreased pup survival, reduced pup weights and malformations (microphthalmia, enophthalmos, domed head) were noted at doses ≥ 15 mg/kg/day (≥ 6.4-times the clinical AUC at the MRHD of 61 mg tafamidis per day). Decreased pup weights in males were associated with delayed sexual maturation (preputial separation) at 15 mg/kg/day. Impaired performance in a water-maze test for learning and memory was observed at 15 mg/kg/day. The no observable adverse effect level (NOAEL) for viability and growth in the F1 generation offspring following maternal exposures to tafamidis was 5 mg/kg/day (human equivalent dose of 0.8 mg/kg/day), a dose approximately 0.92-times the clinical dose of 61 mg tafamidis for a 70 kg adult.
Use in pregnancy. (Category D)
Women of childbearing potential. Contraceptive measures should be used by women of childbearing potential during treatment with Vyndamax, and, due to the prolonged half-life, for one month after stopping treatment.
Vyndamax is not recommended in women of childbearing potential not using contraception.
Pregnancy. There are no adequate data on the use of Vyndamax in pregnant women. Based on findings from animal studies, Vyndamax may cause fetal harm when administered to pregnant women (see above). The potential risk for humans is unknown. Vyndamax is not recommended during pregnancy.
To monitor outcomes of pregnant women exposed to Vyndamax, a tafamidis enhanced surveillance for pregnancy outcomes (TESPO) program has been established. If a pregnancy occurs in a woman being treated with Vyndamax, medical or healthcare professionals are encouraged to report the pregnancy to the Sponsor (see Section 8 Sponsor).
Use in lactation. Nonclinical data demonstrate that tafamidis is secreted in the milk of lactating rats. While the effect of Vyndamax on nursing infants after administration to the mother has not been studied, a risk to infants cannot be excluded. Breastfeeding is not recommended during treatment with Vyndamax.
4.7 Effects on Ability to Drive and Use Machines
No studies on the effects of Vyndamax on the ability to drive or use machines have been performed.
4.8 Adverse Effects (Undesirable Effects)
The data across clinical trials reflect exposure of 377 ATTR-CM patients to either 20 mg or 80 mg (administered as four 20 mg capsules) of tafamidis meglumine daily for an average of 24.5 months (ranging from 1 day to 111 months). Tafamidis 61 mg (Vyndamax) was not administered in the pivotal study (see Section 5.2 Pharmacokinetic Properties). The population included adult patients diagnosed with ATTR-CM, the majority (approximately 90%) of whom had a baseline NYHA (New York Heart Association) classification of Class II or Class III. The mean age was approximately 75 years (ranging from 46 years to 91 years of age); a majority were male (> 90%), and approximately 82% were Caucasian.
Adverse events were assessed from ATTR-CM clinical trials with tafamidis meglumine including a 30-month placebo-controlled trial in patients diagnosed with ATTR-CM (see Section 5.1 Pharmacodynamic Properties). The frequency of adverse events in patients treated with 20 mg or 80 mg tafamidis meglumine was similar and comparable to placebo.
The most commonly reported treatment-emergent adverse events (> 15%) for the patients treated with tafamidis meglumine (pooled tafamidis meglumine 20 mg + 80 mg) compared to placebo in the pivotal phase 3 Study B3461028 are shown in Table 1.
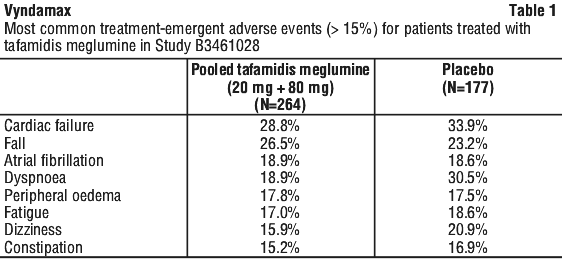
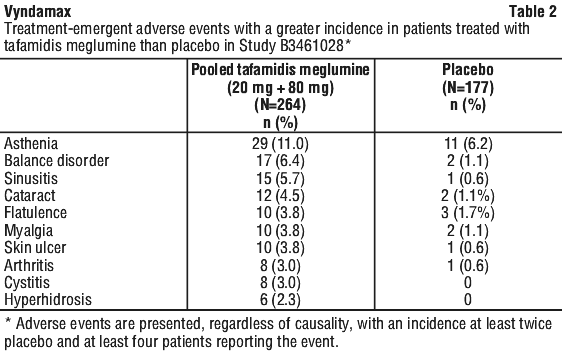
Incidence of hypothyroidism was reported in 6.8%, 5.7% and 5.6% patients in the tafamidis meglumine 80 mg, 20 mg and placebo groups, respectively (see Section 4.4 Special Warnings and Precautions for Use).
Liver function test increased were more frequent in the tafamidis meglumine 80 mg group (3.4%) than in the tafamidis meglumine 20 mg (2.3%) and placebo (1.1%) groups. A causal relationship has not been established (see Section 4.4 Special Warnings and Precautions for Use).
Low neutrophil count (< 0.8 x LLN) was more frequent with tafamidis meglumine treatment than with placebo (1.9% tafamidis meglumine 80 mg, 1.2% tafamidis meglumine 20 mg, 0.6% placebo).
Post-marketing experience. The adverse drug reactions (ADRs) listed below are identified post-marketing, and are presented by MedDRA system organ class (SOC) and frequency categories, defined using the following convention: very common (≥ 10%), common (≥ 1% to < 10%), uncommon (≥ 0.1% to < 1%), rare (≥ 0.01% to < 0.1%) or very rare (< 0.01%).
Gastrointestinal disorders. Common: diarrhoea.
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
4.9 Overdose
Symptoms. There is minimal clinical experience with overdose. During clinical trials, two patients diagnosed with ATTR-CM accidentally ingested a single tafamidis meglumine dose of 160 mg without the occurrence of any associated adverse events. The highest dose of tafamidis meglumine given to healthy volunteers in a clinical trial was 480 mg as a single dose. There was one reported treatment-related adverse event of mild hordeolum at this dose.
Management. In case of overdose, standard supportive measures should be instituted as required.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Other nervous system drugs, ATC code: N07XX08.
Mechanism of action. Tafamidis is a selective stabiliser of TTR. Tafamidis binds with negative cooperativity to the two thyroxine binding sites on the native tetrameric form of TTR preventing dissociation into monomers, the rate-limiting step in the amyloidogenic process. The inhibition of TTR tetramer dissociation forms the rationale for the use of Vyndamax to reduce all-cause mortality and cardiovascular-related hospitalisation in ATTR-CM patients.
No studies have been undertaken to establish a direct relationship between this dissociation and an effect on reduction of amyloid deposition in the heart.
Pharmacodynamic effects. A TTR stabilisation assay was utilised as a pharmacodynamic marker and assessed the stability of the TTR tetramer under denaturation conditions. The TTR stabilisation assay quantifies immunoturbidimetric measurement of the stable TTR tetramer in plasma pre- and post-treatment with 2-day in vitro denaturation with urea. Using this assay, a dose-dependent trend for greater TTR tetramer stabilisation is observed for tafamidis meglumine 80 mg compared to tafamidis meglumine 20 mg. However, the clinical relevance of a higher TTR tetramer stabilisation towards cardiovascular outcomes is not known.
Tafamidis meglumine stabilised both the wild-type TTR tetramer and the tetramers of 14 TTR variants tested clinically after once-daily dosing. Tafamidis meglumine also stabilised the TTR tetramer for an additional 25 variants tested ex vivo, thus demonstrating TTR stabilisation of 40 amyloidogenic TTR genotypes.
A population PK/PD analysis was conducted with a database consisting of 3,662 observations from 102 healthy subjects and 558 patients with transthyretin amyloidosis.
None of the following parameters were found to modify the tafamidis meglumine pharmacodynamic response: race (non-Japanese vs. Japanese), patient type, or genotype.
Clinical trials. Efficacy was demonstrated in a multicentre, international, double-blind, placebo-controlled, randomised 3-arm study in 441 patients with wild-type or hereditary ATTR-CM.
Eligible patients were between 18 and 90 years old at the time of randomisation with a medical history of heart failure and documented ATTR-CM (variant or wild-type). Patients had documented presence of amyloid deposits in biopsy tissue and demonstration of TTR precursor protein at screening or previously, end-diastolic intraventricular septal wall thickness > 12 mm by echocardiography, N-terminal prohormone B-type natriuretic peptide (NT-proBNP) ≥ 600 picogram/mL and 6-Minute Walk Test (6MWT) > 100 m at screening. Patients with NYHA Class I-III were enrolled, whilst patients with NYHA Class IV or transplant patients were excluded.
Patients were randomised to either tafamidis meglumine 20 mg (n=88) or 80 mg [administered as four 20 mg tafamidis meglumine capsules] (n=176) or matching placebo (n=177) once daily, in addition to standard of care (e.g. diuretics) for 30 months. Treatment assignment was stratified by the presence or absence of a variant TTR genotype as well as by baseline severity of disease (NYHA Class).
Tafamidis 61 mg (Vyndamax) was not administered in the pivotal study (see Section 5.2 Pharmacokinetic Properties).
Table 3 describes the patient demographics and baseline characteristics.
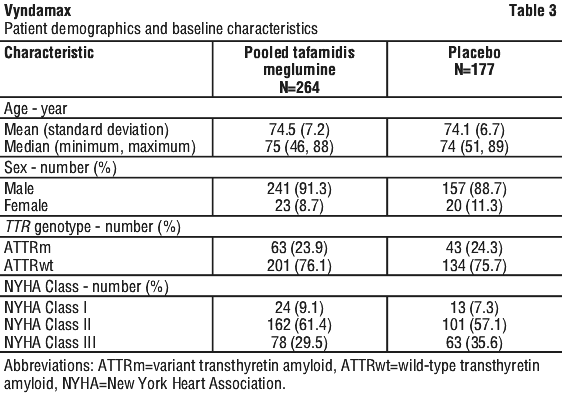
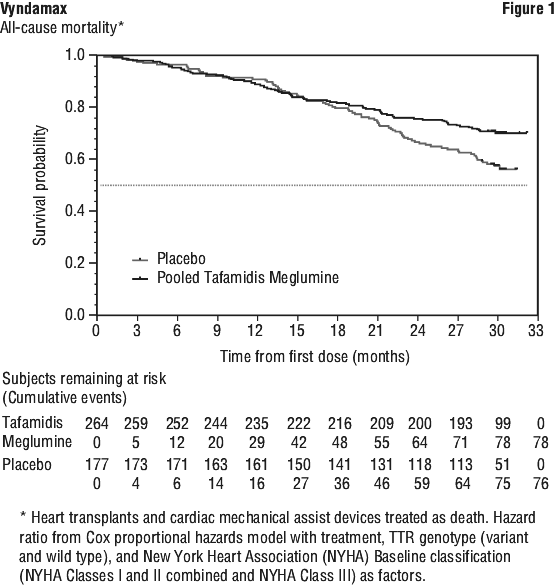
This analysis demonstrated a significant reduction (p=0.0006) in all-cause mortality and frequency of cardiovascular-related hospitalisations in the pooled tafamidis meglumine 20 mg and 80 mg dose group versus placebo (Table 4).

The hazard ratio from the all-cause mortality Cox-proportional hazard model for pooled tafamidis meglumine was 0.698 (95% CI 0.508, 0.958), indicating a 30.2% reduction in the risk of death relative to the placebo group (p=0.0259). Approximately 80% of total deaths were cardiovascular-related in both treatment groups. A Kaplan-Meier plot of time to event all-cause mortality is presented in Figure 1.
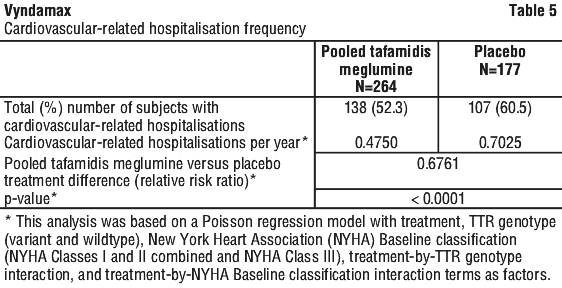
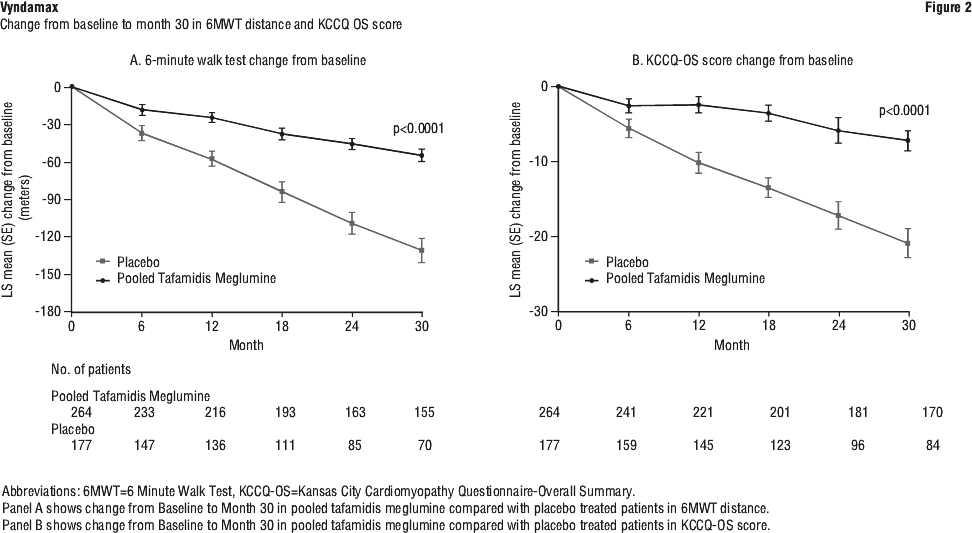
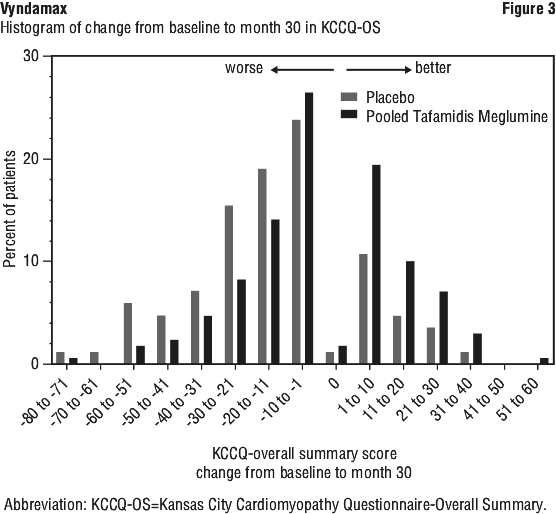

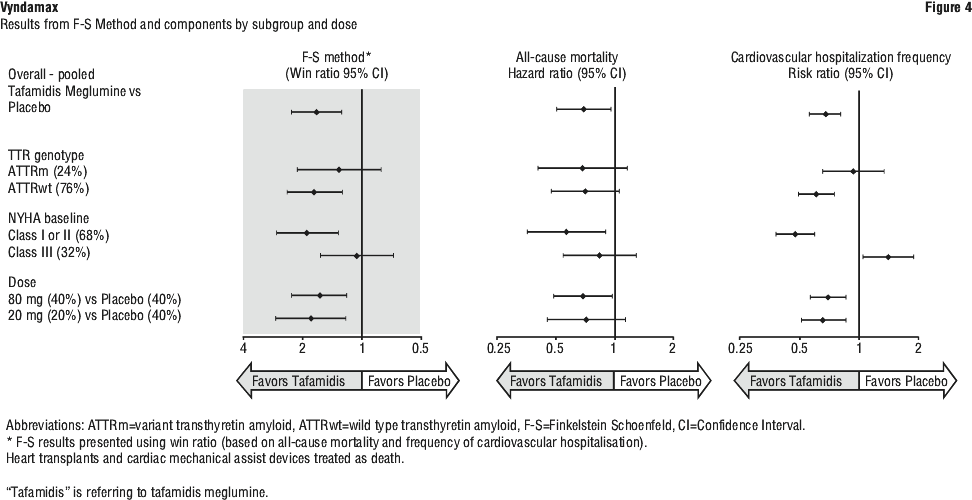
Results from F-S method represented by win ratio for the combined endpoint and its components (all-cause mortality and frequency of cardiovascular-related hospitalisation) consistently favoured tafamidis meglumine versus placebo across all subgroups (wild-type, variant and NYHA Class I, II, and III) except for cardiovascular-related hospitalisation frequency in NYHA Class III (Figure 4) which is higher in the tafamidis meglumine group versus placebo. Win ratio is the number of pairs of treated-patient "wins" divided by number of pairs of placebo patient "wins". Analyses of 6MWT and KCCQ-OS also favoured tafamidis meglumine relative to placebo within each subgroup.
Biomarkers associated with heart failure (NT-proBNP and Troponin I) differentiated between the 80 mg and the 20 mg doses. For NT-proBNP, the LS mean difference in change from Baseline to Month 30 from placebo for 20 mg tafamidis meglumine was -1417.02 picogram/mL (SE=743.38) and for 80 mg was -2587.54 picogram/mL (SE=570.25). Further, the LS mean difference between the 20 mg and 80 mg doses was 1170.51 picogram/mL (SE=587.31) (p=0.0468), favouring the 80 mg dose group. Similar results were observed for Troponin I where the LS mean difference in change from Baseline to Month 30 from placebo for 20 mg tafamidis meglumine was -0.06 nanogram/mL (SE=0.045) and for 80 mg was -0.10 nanogram/mL (SE=0.018). The LS mean difference between the 20 mg and 80 mg doses for Troponin I was 0.05 nanogram/mL (SE=0.04) (p=0.2479), favouring the 80 mg dose group numerically, but not statistically.
A supra-therapeutic, single, 400 mg oral dose of tafamidis meglumine solution in healthy volunteers demonstrated no effect on the QTc interval.
5.2 Pharmacokinetic Properties
The pharmacokinetic profile of tafamidis meglumine was determined in Phase I studies in healthy volunteers and patients with ATTR-CM.
Absorption. After oral administration of Vyndamax once daily, the maximum peak concentration (Cmax) is achieved at a median time (tmax) within 4 hours after dosing in the fasted state. Concomitant administration of a high fat, high calorie meal altered the rate of absorption, but not the extent of absorption. These results support the administration of Vyndamax with or without food.
Distribution. Tafamidis is highly protein bound (> 99%) in plasma. The apparent steady-state volume of distribution is 18.5 litres in a 75 kg adult.
Metabolism and excretion. While there is no explicit evidence of biliary excretion of tafamidis in humans, based on preclinical data, it is suggested that tafamidis is metabolised by glucuronidation and excreted via the bile. This route of metabolism and excretion is likely in humans, as approximately 59% of the total administered dose is recovered in faeces mostly as unchanged drug, and approximately 22% recovered in urine mostly as the glucuronide metabolite. Based on population pharmacokinetic results, the apparent oral clearance of tafamidis meglumine is 0.228 L/h (0.263 L/h for tafamidis) and the population mean half-life is approximately 49 hours.
Exposure from once-daily dosing with tafamidis meglumine increased with increasing dose up to 480 mg single dose and multiple doses up to 80 mg/day. In general, increases were proportional or near proportional to dose.
Mean half-life and oral clearance were similar after single and repeated administration of a 20 mg dose of tafamidis meglumine, indicating a lack of induction or inhibition of tafamidis metabolism.
Results of once-daily dosing with tafamidis meglumine 15 mg to 60 mg oral solution for 14 days demonstrated that steady-state (ss) was achieved by Day 14.
Comparative pharmacokinetics. Tafamidis 61 mg provides steady-state exposures (Cmax and AUC) equivalent to 80 mg tafamidis meglumine (administered as four 20 mg capsules), which was administered to patients with ATTR-CM in the double-blind, placebo-controlled, randomised study (Table 7) (see Section 5.1 Pharmacodynamic Properties).
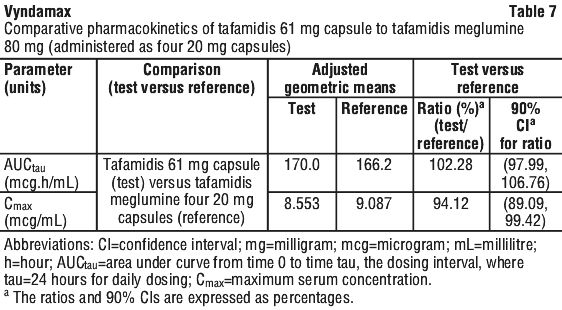
The exposure to Vyndamax in patients with severe hepatic impairment is unknown.
Renal impairment. Vyndamax has not specifically been evaluated in patients with renal impairment. Tafamidis is primarily metabolised by glucuronidation and is likely excreted via the hepatobiliary pathway. The influence of creatinine clearance on tafamidis pharmacokinetics (PK) was evaluated in a population PK analysis in patients with creatinine clearance > 18 mL/min. Pharmacokinetic estimates indicated no difference in apparent oral clearance of tafamidis in patients with creatinine clearance < 80 mL/min compared to those with creatinine clearance ≥ 80 mL/min. No dosage adjustment is required in patients with renal impairment. Limited data are available in patients with severe renal impairment (creatinine clearance ≤ 30 mL/min).
Elderly. Based on population pharmacokinetic results, patients ≥ 65 years of age had an average 15% lower estimate of apparent oral clearance at steady-state when compared with patients under the age of 65. However, the difference in clearance results in < 20% increases in mean Cmax and AUC compared to younger subjects and is not clinically significant.
5.3 Preclinical Safety Data
Genotoxicity. Tafamidis was not genotoxic in a bacterial reverse mutation assay, in an in vitro human lymphocyte chromosomal aberration assay or in an in vivo rat bone marrow micronucleus test. Nonclinical data demonstrated no special hazard for humans based on conventional studies of genotoxicity.
Carcinogenicity. There was no evidence of increased incidence of neoplasia in a 2-year carcinogenicity study in rats at exposures up to 18-times the human AUC at the clinical dose of 61 mg tafamidis. There was no evidence of an increased incidence of neoplasia in the transgenic (Tg)-rasH2 mouse following repeat daily administration for 26 weeks at exposures up to 9.6-times the human AUC at the clinical doses of 61 mg tafamidis. In this study, significant non-neoplastic lesions were noted in the kidneys (nephrosis) and liver (centrilobular hypertrophy and single cell necrosis) in the Tg-rasH2 mice at dose levels ≥ 2.8-times the clinical AUC of 61 mg tafamidis.
6 Pharmaceutical Particulars
6.1 List of Excipients
Macrogol 400, polysorbate 20, povidone, butylated hydroxytoluene.
Gelatin shell (117781). Gelatin, glycerol, iron oxide red, partially dehydrated liquid sorbitol, sorbitol, mannitol.
Opacode WB water based monogramming ink NSP-78-18022 white (PI 3883). Ethanol, isopropyl alcohol, macrogol 400, polyvinyl acetate phthalate, propylene glycol, titanium dioxide, strong ammonium solution.
6.2 Incompatibilities
See Section 4.5 Interactions with Other Medicines and Other Forms of Interactions.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Do not store above 25°C.
6.5 Nature and Contents of Container
Not all presentations may be available.
Cartons containing Aluminium/Aluminium blisters of 30 soft capsules.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
7 Medicine Schedule (Poisons Standard)
Schedule 4.
Date of First Approval
16 March 2020
Date of Revision
15 January 2026
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.