Zejula Capsules
Brand Information
| Brand name | Zejula Capsules |
| Active ingredient | Niraparib |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Zejula Capsules.
Summary CMI
ZEJULA Tablets
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
▼ This medicine is new or being used differently. Please report side effects. See the full CMI for further details.
1. Why am I using ZEJULA?
ZEJULA contains the active ingredient niraparib. ZEJULA is used as a treatment for ovarian, fallopian tube or peritoneal cancer that has completely or partially responded to treatment with platinum-based chemotherapy.
For more information, see Section 1. Why am I using Zejula? in the full CMI.
2. What should I know before I use ZEJULA?
Do not use ZEJULA if you have ever had an allergic reaction to any medicine containing niraparib, lactose or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use Zejula? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with ZEJULA and affect how it works. A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use ZEJULA?
Follow dosage directions given to you by your doctor or pharmacist and continue taking it for as long as your doctor tells you. More instructions can be found in Section 4. How do I use Zejula? in the full CMI.
5. What should I know while using ZEJULA?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using Zejula? in the full CMI.
6. Are there any side effects?
The following common side effects may occur when taking ZEJULA: tiredness, weakness, nausea, stomach pain, vomiting, constipation, diarrhoea, indigestion, decreased appetite, inability to sleep, headache, dizziness, runny or stuffy nose, shortness of breath, cough, high blood pressure, urinary tract infection, heart palpitations, back pain and joint pain. If you experience bruising and bleeding for longer than usual, fever or infection, or shortness of breath alongside tiredness, pale skin and a fast heartbeat or a life-threatening allergic reaction then contact your doctor immediately. For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
▼ This medicine is subject to additional monitoring. This will allow quick identification of new safety information. You can help by reporting any side effects you may get. You can report side effects to your doctor, or directly at www.tga.gov.au/reporting-problems.
ZEJULA Tablets
Active ingredient(s): Niraparib
Consumer Medicine Information (CMI)
This leaflet provides important information about using ZEJULA. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using ZEJULA.
Where to find information in this leaflet:
1. Why am I using Zejula?
2. What should I know before I use Zejula?
3. What if I am taking other medicines?
4. How do I use Zejula?
5. What should I know while using Zejula?
6. Are there any side effects?
7. Product details
1. Why am I using ZEJULA?
ZEJULA contains the active ingredient niraparib.
Niraparib is a type of anti-cancer medicine called a PARP inhibitor. PARP inhibitors block an enzyme called poly [adenosine diphosphate-ribose] polymerase (PARP).
PARP helps cells repair damaged DNA, so blocking it means that the DNA of cancer cells cannot be repaired. This results in tumour cell death, helping to control the cancer.
ZEJULA is used in adults for the treatment of cancer of the ovary, the fallopian tubes (part of the female reproductive system that connects the ovaries to the uterus), or the peritoneum (the membrane lining the abdomen).
It is used for the treatment of cancer that has:
- responded to the first treatment with platinum-based chemotherapy, or
- come back (recurred) after the cancer has responded to previous treatment with standard platinum-based chemotherapy.
Ask your doctor if you have any questions about why it has been prescribed for you.
Your doctor may have prescribed it for another purpose.
This medicine is available only with a doctor's prescription.
There is not enough information to recommend the use of this medicine for children under the age of 18 years.
2. What should I know before I use ZEJULA?
Warnings
Always check the ingredients to make sure you can use this medicine.
Do not use ZEJULA if:
- you are allergic to niraparib, or any of the ingredients listed at the end of this leaflet. Some symptoms of an allergic reaction include: skin rash, itching, shortness of breath or swelling of the face, lips or tongue, which may cause difficulty in swallowing or breathing.
- you are pregnant, intend to become pregnant or are breastfeeding.
Check with your doctor if you:
- have any other medical conditions including low blood counts, high blood pressure, myelodysplatic syndrome (MDS)/acute myeloid leukaemia (AML) or posterior reversible encephalopathy syndrome (PRES).
- take any medicines for any other condition
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Do not take ZEJULA if you are breastfeeding.
It is not known if ZEJULA passes into breast milk. If you are breastfeeding, you must stop before you start taking ZEJULA and you must not begin breast-feeding until 1 month after taking your last dose.
Do not take this medicine if you are pregnant or intend to become pregnant.
ZEJULA can harm your unborn baby and may cause loss of pregnancy (miscarriage).
If you are a woman who could become pregnant you must use highly effective contraception while you are taking ZEJULA, and you must continue to use reliable contraception for 6 months after taking your last dose.
If you are able to become pregnant, your doctor may perform a pregnancy test before you start treatment with ZEJULA.
Contact your doctor straightaway if you become pregnant while you are taking ZEJULA.
Medical Conditions
Tell your doctor or pharmacist if you have or have had any of the following medical conditions:
- Low blood counts
ZEJULA lowers your blood-cell counts, such as your red blood-cell count (anaemia), white blood-cell count (neutropaenia), or blood-platelet count (thrombocytopaenia). Signs and symptoms you need to look out for include fever or infection, and abnormal bruising or bleeding. Your doctor will test your blood regularly throughout your treatment.
- Myelodysplastic syndrome (MDS)/acute myeloid leukaemia (AML)
Rarely, low blood-cell counts may be a sign of more serious problems with the bone marrow such as ‘myelodysplastic syndrome’ (MDS) or ‘acute myeloid leukaemia’ (AML). Your doctor may want to test your bone marrow to check for these problems.
- High blood pressure
ZEJULA can cause high blood pressure, which in some cases, could be severe. Your doctor will measure your blood pressure regularly throughout your treatment. He or she may also give you medicine to treat high blood pressure and adjust your ZEJULA dose, if necessary.
- Posterior Reversible Encephalopathy Syndrome (PRES)
A rare neurological side effect named Posterior Reversible Encephalopathy Syndrome (PRES) has been associated with ZEJULA treatment. If you have headache, vision changes, confusion or seizure with or without high blood pressure, please contact your doctor.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and ZEJULA may interfere with each other. These include:
- medicines used to prevent rejection after an organ transplant, such as cyclosporin and tacrolimus
- alfentanil, a medicine used to manage pain
- ergotamine, a medicine to treat migraine
- medicines used to treat mental disorders such as pimozide, quetiapine and clozapine
- halofantrine, a medicine to treat malaria
- theophylline, a medicine to treat asthma
- ropinirole, a medicine to treat Parkinson's disease
- irinotecan, a medicine to treat cancer
- medicines to treat high cholesterol such as rosuvastatin, simvastatin and atorvastatin
- methotrexate, a medicine used to treat cancer, rheumatoid arthritis or psoriasis
- metformin, a medicine used to treat diabetes
These medicines may be affected by ZEJULA or may affect how well it works. You may need to use different amounts of your medicine or take different medicines.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect ZEJULA
4. How do I use ZEJULA?
Follow all directions given to you by your doctor or pharmacist carefully.
They may differ from the information contained in this leaflet.
If you do not understand the instructions on the pack, ask your doctor or pharmacist for help.
Do not change the ZEJULA dose without talking to your doctor.
How much to take
The recommended starting dose is 2 tablets taken together once a day (total daily dose of 200 mg). For some patients, a starting dose of 300 mg (3 tablets) may be appropriate and recommended by your doctor based on clinical assessment.
Your doctor may recommend a lower dose if you experience side effects (such as nausea, tiredness, abnormal bleeding/bruising, anaemia) or if you have problems with your liver.
Follow the instructions provided and use ZEJULA until your doctor tells you to stop.
When to take ZEJULA
Take ZEJULA at approximately the same time each day.
Taking ZEJULA at bedtime may help you to manage nausea.
How to take ZEJULA
Swallow ZEJULA whole with a glass of water. Do not open, chew or crush the tablets.
It does not matter if you take this medicine with or without food.
Continue taking your medicine for as long as your doctor tells you. Your doctor will check you on a regular basis, and you will normally continue to take Zejula until disease progression, and as long as you do not suffer unacceptable side effects.
If you forget to take ZEJULA
ZEJULA should be used regularly at the same time each day. If you miss your dose at the usual time, take your next dose at its scheduled time. Do not take an additional dose if you a miss a dose or vomit after taking ZEJULA.
Do not take a double dose to make up for the dose you missed. This may increase the chance of getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you are having trouble remembering when to take your medicine, ask your doctor or pharmacist for hints.
If you use too much ZEJULA
If you think that you have used too much ZEJULA, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using ZEJULA?
Things you should do
- If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking ZEJULA.
- If you are a woman who could become pregnant you must use highly effective contraception while you are taking ZEJULA, and you must continue to use reliable contraception for 6 months after taking your last dose.
- Keep all of your doctor's appointments so that your progress can be checked.
Your doctor may do some tests from time to time to make sure the medicine is working and to prevent unwanted side effects. Your doctor will test your blood weekly for the first month, then monthly for 10 months and afterwards periodically
Call your doctor straight away if you:
- Become pregnant while taking this medicine.
Remind any doctor, dentist or pharmacist you visit that you are using ZEJULA.
Things you should not do
- Do not use this medicine to treat any other complaints unless your doctor tells you to.
- Do not give this medicine to anyone else, even if they have the same condition as you.
- Do not stop taking ZEJULA or lower the dosage without checking with your doctor.
If you stop taking it suddenly, your condition may worsen. Your doctor may interrupt your treatment or reduce your dose if you are having unwanted side effects.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how ZEJULA affects you.
ZEJULA may cause dizziness, tiredness, difficulty concentrating or weakness in some people. If you have any of these symptoms, do not drive, operate machinery or do anything else that could be dangerous.
Looking after your medicine
- Keep your tablets in the pack until it is time to take them. If you take the tablets out of the pack they may not keep well.
- Keep ZEJULA in a cool, dry place where the temperature stays below 30°C.
Follow the instructions in the carton on how to take care of your medicine properly.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on windowsills.
Keep it where young children cannot reach it.
A locked cupboard at least one-and-half metres above the ground is a good place to store medicines.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
General disorders:
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
Blood Related:
Some other serious side effects may only become known through tests | Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What ZEJULA contains
| Active ingredient (main ingredient) | Niraparib (as niraparib tosilate monohydrate) |
| Other ingredients inactive ingredients) | Microcrystalline cellulose Lactose monohydrate Povidone Crospovidone Silicon dioxide Magnesium stearate Polyvinyl alcohol Titanium dioxide Macrogol 4000 Purified talc Ferrosoferric oxide |
Do not take this medicine if you are allergic to any of these ingredients.
What ZEJULA looks like
Zejula 100 mg tablets are immediate release, grey, oval-shaped, film coated tablets, debossed with “100” on one side and "Zejula" on the other side.
Zejula is available in packs of 56 and 84 tablets (AUST R 395406).
Who distributes ZEJULA
GlaxoSmithKline Australia Pty Ltd
Level 4, 436 Johnston Street,
Abbotsford, Victoria, 3067
Phone: 1800 033 109
www.gsk.com.au
©2023 GSK group of companies or its licensor.
This leaflet was prepared in September 2023.
Version 2.0
Brand Information
| Brand name | Zejula Capsules |
| Active ingredient | Niraparib |
| Schedule | S4 |
▼ This medicinal product is subject to additional monitoring in Australia. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse events at www.tga.gov.au/reporting-problems.
MIMS Revision Date: 01 March 2026
1 Name of Medicine
Zejula (niraparib).
2 Qualitative and Quantitative Composition
Each capsule contains niraparib tosilate monohydrate equivalent to 100 mg niraparib. Excipients with known effect: Each capsule contains lactose monohydrate. Each capsule shell also contains the colouring agent tartrazine.
Each film-coated tablet contains niraparib tosilate monohydrate equivalent to 100 mg. Excipients with known effect: Each film-coated tablet contains lactose monohydrate.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Capsule. Hard capsules have a white body with "100 mg" printed in black ink and purple cap with "Niraparib" printed in white ink.
Tablets. 100 mg grey, oval-shaped, film-coated tablet debossed with "100" on one side and "Zejula" on the other.
4 Clinical Particulars
4.1 Therapeutic Indications
Zejula is indicated:
for the maintenance treatment of adult patients with advanced high-grade ovarian, fallopian tube or primary peritoneal cancer who are in response (complete or partial) following completion of first-line platinum-based chemotherapy;
as monotherapy for the maintenance treatment of adult patients with platinum-sensitive relapsed high-grade serous epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in response (complete or partial) to platinum-based chemotherapy.
4.2 Dose and Method of Administration
Treatment with Zejula should be initiated and supervised by a physician experienced in the use of anticancer medicinal products.
First-line ovarian cancer maintenance treatment. The recommended starting dose of niraparib is 200 mg (two 100 mg capsules or tablets) taken once daily. For patients who weigh ≥ 77 kg and have baseline platelet count ≥ 150,000/microL, the recommended starting dose of niraparib is 300 mg (three 100 mg capsules or tablets) taken orally once daily.
Recurrent ovarian cancer maintenance treatment. The recommended starting dose is 300 mg (three 100 mg capsules or tablets) taken orally once daily.
Patients should be encouraged to take their dose at approximately the same time each day. Bedtime administration may be a potential method for managing nausea.
The capsules or tablets should be swallowed whole with water. The capsules or tablets should not be chewed or crushed. Zejula can be taken without regard to meals (see Section 5.2 Pharmacokinetic Properties).
It is recommended that treatment should be continued until disease progression or unacceptable toxicity.
Missing dose. If patients miss a dose, they should take their next dose at its regularly scheduled time.
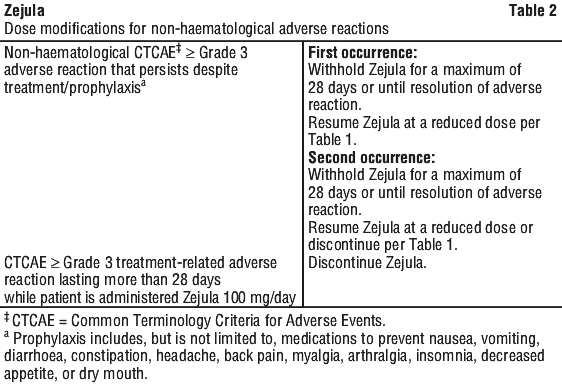
Dose adjustments for adverse reactions. To manage adverse reactions, consider interruption of treatment, dose reduction, or dose discontinuation. The recommended dose modifications for adverse reactions are listed in Tables 1, 2 and 3.


Renal impairment. No dose adjustment is necessary for patients with mild to moderate renal impairment. There are no data in patients with severe renal impairment or end stage renal disease undergoing haemodialysis; use with caution in these patients, see Section 5.2 Pharmacokinetic Properties.
Hepatic impairment. No dose adjustment is needed in patients with mild hepatic impairment.
For patients with moderate hepatic impairment, the recommended starting dose of Zejula is 200 mg once daily, see Section 5.2 Pharmacokinetic Properties.
There are no data in patients with severe hepatic impairment; use with caution in these patients, see Section 5.2 Pharmacokinetic Properties.
Patients with ECOG performance status 2 to 4. Clinical data are not available in patients with ECOG performance status 2 to 4.
Paediatric population. The safety and efficacy of Zejula in children and adolescents below 18 years of age have not yet been established. No data are available.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of Excipients.
Breastfeeding (see Section 4.6 Fertility, Pregnancy and Lactation).
4.4 Special Warnings and Precautions for Use
Haematological adverse reactions. Haematological adverse reactions (thrombocytopenia, anaemia, neutropenia) have been reported in patients treated with niraparib. In the PRIMA and NOVA studies, patients eligible for Zejula therapy had the following baseline haematological parameters: absolute neutrophil count (ANC) ≥ 1,500 cells/microL; platelets ≥ 100,000 cells/microL and haemoglobin ≥ 10 g/dL (PRIMA) or ≥ 9 g/dL (NOVA) prior to therapy.
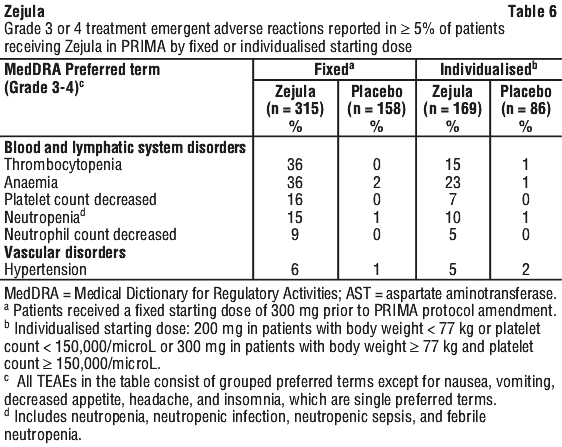
In the PRIMA study, the overall incidence of Grade ≥ 3 thrombocytopenia, anaemia and neutropenia in clinical and/or laboratory findings were reported in 39%, 31%, and 21% of patients receiving Zejula. Discontinuation due to thrombocytopenia, anaemia, and neutropenia occurred in 4%, 2%, and 2% of patients, respectively.
In patients who were administered a starting dose of Zejula based on baseline weight or platelet count, Grade ≥ 3 thrombocytopenia, anaemia and neutropenia were reported in 15%, 23%, and 10% of patients receiving Zejula, respectively. Discontinuation due to thrombocytopenia, anaemia, and neutropenia occurred in 3%, 3%, and 2% of patients, respectively.
In the NOVA study, Grade ≥ 3 thrombocytopenia, anaemia and neutropenia were reported in 29%, 25%, and 20% of patients receiving Zejula, respectively. Discontinuation due to thrombocytopenia, anaemia, and neutropenia occurred in 3%, 1%, and 2% of patients, respectively.
If a patient develops severe persistent haematologic toxicity including pancytopenia that does not resolve within 28 days following interruption, Zejula should be discontinued.
Test complete blood counts weekly for the first month, followed by monthly monitoring for the next 10 months of treatment and periodically after this time to monitor for clinically significant changes in any haematologic parameter during treatment, see Section 4.2 Dose and Method of Administration.
Due to the risk of thrombocytopenia, anticoagulants and medicinal products known to reduce the thrombocyte count should be used with caution, see Section 4.8 Adverse Effects (Undesirable Effects).
Myelodysplastic syndrome/acute myeloid leukaemia. Myelodysplastic syndrome/acute myeloid leukaemia (MDS/AML), including cases with fatal outcome, have been reported in patients who received Zejula (see Section 4.8 Adverse Effects (Undesirable Effects)).
In clinical trials, the duration of Zejula treatment in patients prior to developing MDS/AML varied from 1 month to > 4 years. The cases were typical of secondary, cancer therapy-related MDS/AML. All patients had received platinum-containing chemotherapy regimens and many had also received other DNA damaging agents and radiotherapy. Some of the patients had a history of bone marrow dysplasia.
For suspected MDS/AML or prolonged haematological toxicities, the patient should be referred to a haematologist for further evaluation. If MDS/AML is confirmed, treatment with Zejula, treatment should be discontinued.
Hypertension, including hypertensive crisis. Hypertension, including hypertensive crisis, has been reported with the use of Zejula (see Section 4.8 Adverse Effects (Undesirable Effects)). Pre-existing hypertension should be adequately controlled before starting Zejula treatment. Blood pressure and heart rate should be monitored at least weekly for the first two months, then monthly for the first year and periodically thereafter during treatment with Zejula.
Hypertension should be medically managed with antihypertensive medicinal products as well as adjustment of the Zejula dose (see Section 4.2 Dose and Method of Administration), if necessary. In the clinical programme, blood pressure measurements were obtained on Day 1 of each 28-day cycle while the patient remained on Zejula. In most cases, hypertension was controlled adequately using standard antihypertensive treatment with or without Zejula dose adjustment, see Section 4.2 Dose and Method of Administration. Zejula should be discontinued in case of hypertensive crisis or if medically significant hypertension cannot be adequately controlled with antihypertensive therapy.
Posterior reversible encephalopathy syndrome (PRES). There have been rare reports (0.09% of clinical trial patients) of Zejula-treated patients developing signs and symptoms that are consistent with posterior reversible encephalopathy syndrome (PRES) (see Section 4.8 Adverse Effects (Undesirable Effects)). PRES is a rare neurologic disorder that can present with the following signs and symptoms including seizures, headache, altered mental status, visual disturbance, or cortical blindness, with or without associated hypertension. A diagnosis of PRES requires confirmation by brain imaging, preferably magnetic resonance imaging (MRI). In patients developing PRES, treatment of specific symptoms including control of hypertension is recommended, along with discontinuation of Zejula. The safety of reinitiating niraparib therapy in patients previously experiencing PRES is not known.
Pregnancy/contraception. Zejula should not be used during pregnancy or in women of childbearing potential not willing to use highly effective contraception during therapy and for 6 months after receiving the last dose of Zejula (see Section 4.6 Fertility, Pregnancy and Lactation). A pregnancy test should be performed on all women of childbearing potential prior to treatment.
Lactose. Zejula capsules contain lactose monohydrate. Patients with rare hereditary problems of galactose intolerance, the Lapp lactase deficiency or glucose-galactose malabsorption should not take this medicine.
Tartrazine. This medicinal product contains tartrazine, which may cause allergic reactions.
Use in the elderly. See Section 4.2 Dose and Method of Administration.
Paediatric use. See Section 4.2 Dose and Method of Administration.
Effects on laboratory tests. No data available.
4.5 Interactions with Other Medicines and Other Forms of Interactions
Pharmacodynamic interactions. The combination of Zejula with vaccines or immunosuppressant agents has not been studied.
The data on Zejula in combination with cytotoxic medicinal products are limited. Therefore, caution should be taken if Zejula is used in combination with vaccines, immunosuppressant agents or with other cytotoxic medicinal products.
Pharmacokinetic interactions. No clinical drug interaction studies have been performed with niraparib.
Effect of other medicinal products on niraparib. Niraparib as a substrate of CYPs (CYP1A2 and CYP3A4). Niraparib is a substrate of carboxylesterases (CEs) and UDP-glucuronosyltransferases (UGTs) in vivo. Oxidative metabolism of niraparib is minimal in vivo. No dose adjustment for Zejula is required when administered concomitantly with medicinal products known to inhibit (e.g. itraconazole, ritonavir, and clarithromycin) or induce CYP enzymes (e.g. rifampin, carbamazepine, and phenytoin).
Niraparib as a substrate of efflux transporters (P-gp, BCRP, and MATE1/2). Niraparib is a substrate of P-glycoprotein (P-gp) and Breast Cancer Resistance Protein (BCRP). However, due to its high permeability and bioavailability, the risk of clinically relevant interactions with medicinal products that inhibit these transporters is unlikely. Therefore, no dose adjustment for Zejula is required when administered concomitantly with medicinal products known to inhibit P-gp (e.g. amiodarone, verapamil) or BCRP (e.g. osimertinib, velpatasvir, and eltrombopag).
Niraparib is not a substrate of bile salt export pump (BSEP). The major primary metabolite M1 is not a substrate of P-gp, BCRP, or BSEP. Niraparib is not a substrate of MATE 1 or 2, while M1 is a substrate of both.
Niraparib as a substrate of hepatic uptake transporters (OATP1B1, OATP1B3, and OCT1). Neither niraparib nor M1 is a substrate of organic anion transport polypeptide 1B1 (OATP1B1), 1B3 (OATP1B3), or organic cation transporter 1 (OCT1). No dose adjustment for Zejula is required when administered concomitantly with medicinal products known to inhibit OATP1B1 or 1B3 (e.g. gemfibrozil, ritonavir), or OCT1 (e.g. dolutegravir) uptake transporters.
Niraparib as a substrate of renal uptake transporters (OAT1, OAT3, and OCT2). Neither niraparib nor M1 is a substrate of organic anion transporter 1 (OAT1), 3 (OAT3), and organic cation transporter 2 (OCT2). No dose adjustment for Zejula is required when administered concomitantly with medicinal products known to inhibit OAT1 (e.g. probenecid) or OAT3 (e.g. probenecid, diclofenac), or OCT2 (e.g. cimetidine, quinidine) uptake transporters.
Effect of niraparib on other medicinal products. Inhibition of CYPs (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5). In vitro, neither niraparib nor its inactive major metabolite M1 is a clinically relevant inhibitor of any active substance-metabolising CYP enzymes, namely CYP1A1/2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5.
Induction of CYPs (CYP1A2 and CYP3A4/5). Neither niraparib nor M1 is a clinically relevant CYP3A4 inducer in vitro. In vitro, niraparib weakly induces CYP1A2 at high concentrations and the clinical relevance of this effect cannot be completely ruled out. M1 is not a CYP1A2 inducer. Therefore, caution is recommended when niraparib is combined with active substances the metabolism of which is CYP1A2-dependent and, notably, those having a narrow therapeutic range (e.g. clozapine, theophylline, and ropinirole).
Inhibition of efflux transporters (P-gp, BCRP, BSEP, MRP2 and MATE1/2K). Neither niraparib nor M1 is a clinically relevant inhibitor of P-gp, BSEP or MRP2 and may be a weak BCRP inhibitor based on in vitro data and/or physiologically based pharmacokinetic (PBPK) modelling.
Niraparib is an inhibitor of MATE1/2K with IC50 of 0.18 microM and ≤ 0.14 microM, respectively, M1 does not inhibit MATE1/2K. Simulations using PBPK modelling indicate an expected > 2-fold increase in exposure of metformin when administered with niraparib at 200 mg or 300 mg daily. Close monitoring of glycaemia is recommended when starting or stopping niraparib in patients receiving metformin. A dose adjustment of metformin may be necessary.
Inhibition of uptake transporters (OATP1B1, OATP1B3, OCT1, OAT1, OAT3 and OCT2). Neither niraparib nor M1 is an inhibitor of hepatic uptake transporters OATP1B1 or OATP1B3 and renal uptake transporters OAT1, OAT3 or OCT2 in vitro.
In vitro, niraparib inhibits hepatic uptake transporters OCT1 with an IC50 = 34.4 microM and an interaction is considered possible.
All clinical studies have only been performed in adults.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. There are no clinical data on fertility. While no direct fertility studies were conducted in animals, repeat-dose toxicity studies in rats and dogs showed decreased spermatogenesis, small testes and germ cell depletion in the testes and epididymides at niraparib doses of 20 mg/kg/day and 6 mg/kg/day (0.74- and 0.05-times clinical exposure based on AUC, respectively). There was a trend towards reversibility of these findings 2-4 weeks after dosing was stopped.
Use in pregnancy. (Category D)
There are no or a limited amount of data from the use of Zejula in pregnant women. Animal reproductive and developmental toxicity studies have not been conducted. However, based on its mechanism of action, Zejula could cause embryonic or fetal harm, including embryo-lethal and teratogenic effects, when administered to a pregnant woman. Zejula should not be used during pregnancy.
Women of childbearing potential should not become pregnant while on treatment and should not be pregnant at the beginning of treatment. A pregnancy test should be performed on all women of childbearing potential prior to treatment. Women of childbearing potential must use highly effective contraception during therapy and for 6 months after receiving the last dose of Zejula.
Use in lactation. It is unknown whether niraparib or its metabolites are excreted in human milk. Breastfeeding is contraindicated during administration of Zejula and for 1 month after receiving the last dose (see Section 4.3 Contraindications).
4.7 Effects on Ability to Drive and Use Machines
Zejula may influence the ability to drive or use machines. Patients who take Zejula may experience asthenia, fatigue, difficulty concentrating and dizziness. Patients who experience these symptoms should observe caution when driving or using machines.
4.8 Adverse Effects (Undesirable Effects)
Tabulated list of adverse reactions. The following adverse reactions have been identified based on pooled data generated from the PRIMA and NOVA clinical trials in patients receiving Zejula monotherapy and during post-marketing experience (see Table 4).
Frequencies of occurrence of undesirable effects are defined as: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000). Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness.

The adverse reactions noted in the group of patients who were administered a 200 mg starting dose of niraparib based on baseline weight or platelet count were of similar or lesser frequency compared to the group administered 300 mg. See Section 4.4 Special Warnings and Precautions for Use for specific information regarding frequency of thrombocytopenia, anaemia and neutropenia. In general, the individualised starting dose decreased the incidence of myelosuppression events including anaemia (71% vs 50%) and thrombocytopenia (52% vs 34%) as compared to the fixed starting dose.
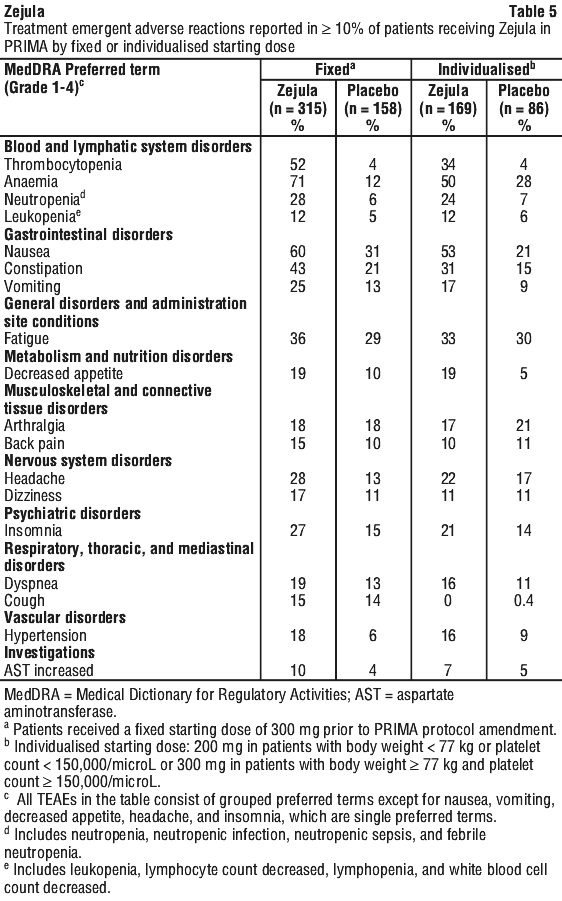
In the clinical programme, haematological adverse reactions were managed with laboratory monitoring and dose modifications (see Section 4.2 Dose and Method of Administration).
Thrombocytopenia. In the PRIMA safety population (n = 484), 39% of Zejula-treated patients experienced Grade 3-4 thrombocytopenia compared to 0.4% of placebo-treated patients with a median time from first dose to first onset in the Zejula arm of 22 days (range: 15 to 335 days) and with a median duration of 6 days (range: 1 to 374 days). Discontinuation due to thrombocytopenia occurred in 4% of patients.
In the NOVA study, approximately 60% of patients receiving Zejula experienced thrombocytopenia of any grade, and 34% of patients experienced Grade 3/4 thrombocytopenia. In patients with baseline platelet count less than 180,000 cells/microL, thrombocytopenia of any grade and Grade 3/4 occurred in 76% and 45% of the patients, respectively. The median time to onset of thrombocytopenia regardless of grade, and Grade 3/4 thrombocytopenia was 22 and 23 days, respectively. The rate of new incidences of thrombocytopenia after intensive dose modifications were performed during the first two months of treatment from Cycle 4 was 1.2%. The median duration of thrombocytopenia events of any grade was 23 days, and the median duration of Grade 3/4 thrombocytopenia was 10 days. Patients treated with Zejula who develop thrombocytopenia might have an increased risk of haemorrhage. Discontinuation due to thrombocytopenia events (thrombocytopenia and platelet count decreased) occurred in approximately 3% of the patients.
In the NOVA study, 48 of 367 (13%) patients experienced bleeding with concurrent thrombocytopenia; all bleeding events concurrent with thrombocytopenia were Grade 1 or 2 in severity except for one event of Grade 3 petechiae and haematoma observed concurrently with a serious adverse event of pancytopenia. Thrombocytopenia occurred more commonly in patients whose baseline platelet count was less than 180,000 cells/microL. Approximately 76% of patients with lower baseline platelets (< 180,000 cells/microL) who received niraparib experienced thrombocytopenia of any grade, and 45% of the patients experienced Grade 3/4 thrombocytopenia. Pancytopenia has been observed in < 1% of patients receiving niraparib.
Anaemia. In the PRIMA safety population (n = 484), 31% of niraparib-treated patients experienced Grade 3/4 anaemia compared to 2% of placebo-treated patients with a median time from first dose to first onset in the niraparib arm of 80 days (range: 15 to 533 days) and with a median duration of 7 days (range: 1 to 119 days). Discontinuation due to anaemia occurred in 2% of patients.
In the NOVA study, approximately 50% of patients experienced anaemia of any grade, and 25% experienced Grade 3/4 anaemia. The median time to onset of anaemia of any grade was 42 days, and 85 days for Grade 3/4 events. The median duration of anaemia of any grade was 63 days, and 8 days for Grade 3/4 events. Anaemia of any grade might persist during Zejula treatment. In the clinical programme, anaemia was managed with laboratory monitoring, dose modification (see Section 4.2 Dose and Method of Administration), and, where appropriate, with red blood cell transfusions. Discontinuation due to anaemia occurred in 1% of patients.
Neutropenia. In the PRIMA safety population (n = 484), 21% of niraparib-treated patients experienced Grade 3-4 neutropenia compared to 1% of placebo-treated patients with a median time from first dose to first onset in the Zejula arm of 29 days (range: 15 to 421 days) and with a median duration of 8 days (range: 1 to 42 days). Discontinuation due to neutropenia occurred in 2% of patients.
In the NOVA study, approximately 30% of patients receiving Zejula experienced neutropenia of any grade, and 20% of patients experienced Grade 3/4 neutropenia. The median time to onset of neutropenia of any grade was 27 days, and 29 days for Grade 3/4 events. The median duration of neutropenia of any grade was 26 days, and 13 days for Grade 3/4 events. In the clinical programme, neutropenia was managed with laboratory monitoring and dose modifications (see Section 4.2 Dose and Method of Administration). In addition, granulocyte-colony stimulating factor (G-CSF) was administered to approximately 6% of patients treated with Zejula as concomitant therapy for neutropenia. Discontinuation due to neutropenia events occurred in 2% of patients.
Myelodysplastic syndrome/acute myeloid leukaemia. In clinical studies, MDS/AML occurred in 1% patients treated with niraparib, with 41% of cases having a fatal outcome. The incidence was higher in patients with relapsed ovarian cancer who had received 2 or more lines of prior platinum chemotherapy and with gBRCAmut following 5.6 years survival follow-up. All patients had potential contributing factors for the development of MDS/AML, having received previous chemotherapy with platinum agents. Many had also received other DNA damaging agents and radiotherapy. The majority of reports were in gBRCAmut carriers. Some of the patients had a history of previous cancer or of bone marrow suppression.
In the PRIMA study, the overall incidence of MDS/AML was 2.3% in patients receiving niraparib and 1.6% in patients received placebo with a follow-up of 6.2 years.
In the NOVA study in patients with relapsed ovarian cancer who had received at least two prior lines of platinum chemotherapy, the overall incidence of MDS/AML was 3.5% in patients receiving niraparib and 1.7% in patients receiving placebo with a follow-up of 5.6 years. In gBRCAmut and non-gBRCAmut cohorts, the incidence of MDS/AML was 6.6% and 1.7% in patients receiving niraparib and 3.1% and 0.9% in patients receiving placebo, respectively.
Hypertension. In the PRIMA study, Grade 3-4 hypertension occurred in 6% of niraparib-treated patients compared to 1% of placebo-treated patients with a median time from first dose to first onset in the Zejula arm of 50 days (range: 1 to 589 days) and with a median duration of 12 days (range: 1 to 61 days). No patients discontinued Zejula due to hypertension.
Hypertension, including hypertensive crisis, has been reported with Zejula therapy.
In the NOVA study, hypertension of any grade occurred in 19.3% of patients treated with Zejula. Grade 3/4 hypertension occurred in 8.2% of patients. Discontinuation due to hypertension occurred in < 1% of patients.
Post-marketing data. Adverse reactions are listed in Table 7 by system organ class and frequency.

4.9 Overdose
There is no specific treatment in the event of Zejula overdose, and symptoms of overdose are not established. In the event of an overdose, physicians should provide general supportive measures and should treat symptomatically.
For information on the management of overdose, contact the Poison Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
ATC code: L01XK02.
Mechanism of action. Niraparib is an inhibitor of poly (ADP-ribose) polymerase (PARP) enzymes, PARP-1 and PARP-2, which play a role in DNA repair. In vitro studies have shown that niraparib-induced cytotoxicity may involve inhibition of PARP enzymatic activity and increased formation of PARP-DNA complexes resulting in DNA damage, apoptosis and cell death. Increased niraparib-induced cytotoxicity was observed in tumour cell lines with or without deficiencies in the BReast CAncer (BRCA) 1 and 2 tumour suppressor genes. In orthotopic high-grade serous ovarian cancer patient derived xenograft tumours (PDX) grown in mice, niraparib has been shown to reduce tumour growth in BRCA 1 and 2 mutant, BRCA wild-type but homologous recombination (HR) deficient, and in tumours that are BRCA wild-type and without detectable HR deficiency.
Clinical trials. First-line ovarian cancer maintenance treatment. PRIMA was a double-blind, placebo-controlled trial in which patients (n = 733) in complete or partial response to first-line platinum-based chemotherapy were randomised 2:1 to niraparib or matched placebo.
Patients were randomised post-completion of first-line platinum-based chemotherapy plus/minus surgery. Bevacizumab was allowed with chemotherapy. Patients who had neoadjuvant chemotherapy followed by interval debulking surgery could have visible residual or no residual disease. Randomisation was stratified by best response during the front-line platinum regimen (complete response vs partial response), neoadjuvant chemotherapy (NACT) (Yes vs No), and homologous recombination deficiency (HR) status [positive vs negative or not determined]. Testing for HRD was performed using the HRD test on tumour tissue obtained at the time of initial diagnosis.
PRIMA was initiated with a starting dose of 300 mg once daily in continuous 28-day cycles (henceforth referred to as a fixed starting dose or FSD). Based on retrospective analyses of the NOVA trial, the starting dose in PRIMA was changed with Amendment 2 of the Protocol. From that point forward, patients with a baseline body weight ≥ 77 kg and baseline platelet count ≥ 150,000/microL were administered niraparib 300 mg (3 x 100 mg capsules) or placebo (3 capsules) daily and patients with a baseline body weight < 77 kg or baseline platelet count < 150,000/microL were administered Zejula 200 mg (2 x 100 mg capsules) or placebo (2 capsules) daily (henceforth referred to as an individualised starting dose or ISD).
Patients began treatment on Cycle 1/Day 1 (C1/D1) with niraparib 200 or 300 mg or matched placebo administered once daily in continuous 28-day cycles. In the PRIMA study, 52% of patients had a dose interruption in Cycle 1, 9% of patients in Cycle 1 and 47% of patients in Cycle 2 had a dose reduction.
Overall, the median dose intensity in subjects who received niraparib was 181.3 mg/day and the median relative dose intensity was 63% in subjects who received niraparib. In patients who received the individualised starting dose, the median dose intensity was 178.6 mg/day and the median relative dose intensity was 66%. In patients who received the fixed starting dose, the median dose intensity was 181.8 mg/day and the median relative dose intensity was 61%.
The major efficacy outcome measure, progression-free survival (PFS), was determined by blinded independent central review (BICR) per RECIST, version 1.1. PFS testing was performed hierarchically: first in the HR-deficient (HRd) population, then in the overall population. Overall survival (OS) was a key secondary endpoint. The median age was 62 and ranged from 32 to 85 years among patients randomised to niraparib and 33 to 88 years among patients randomised to placebo. Eighty-nine percent of all patients were white. Sixty-nine percent of patients randomised with niraparib and 71% of patients randomised with placebo had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 at study baseline. In the overall population, 65% of patients had stage III disease, 35% had stage IV disease and the proportions of patients by primary tumour were as follows: ovarian (80.4%), primary peritoneal (6.4%) and fallopian (13.2%). Most patients (95.5%) had serous tumours and 2.3% of patients had an endometrioid tumour. Sixty-seven percent of the patients received NACT. Sixty-nine percent of the patients had a complete response to the first-line platinum-based chemotherapy.
In the PRIMA study bevacizumab was received by 6 (1.2%) patients in niraparib arm and 1 (0.4) in placebo group. Subjects who had received bevacizumab with their first-line platinum-based therapy but were unable to receive bevacizumab as maintenance therapy due to AEs or for any other reason, were deemed eligible to enter the PRIMA study.
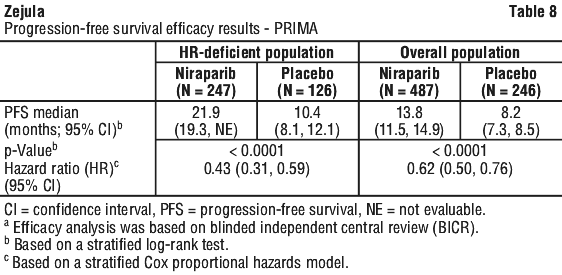
PRIMA demonstrated a statistically significant improvement in PFS for patients randomised to niraparib as compared with placebo in the HR deficient and overall population (Table 8 and Figures 1 and 2).
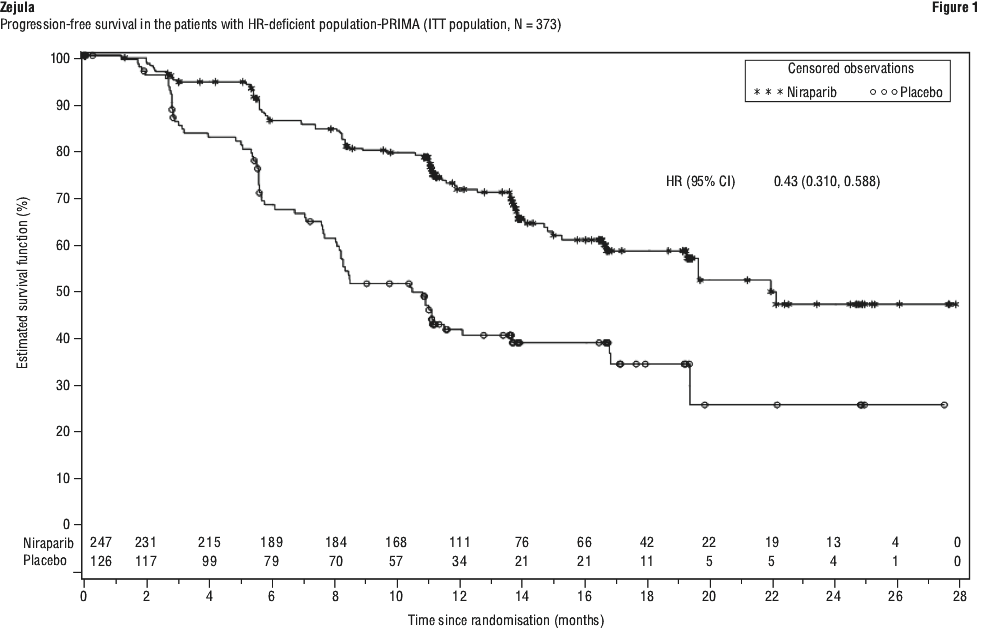
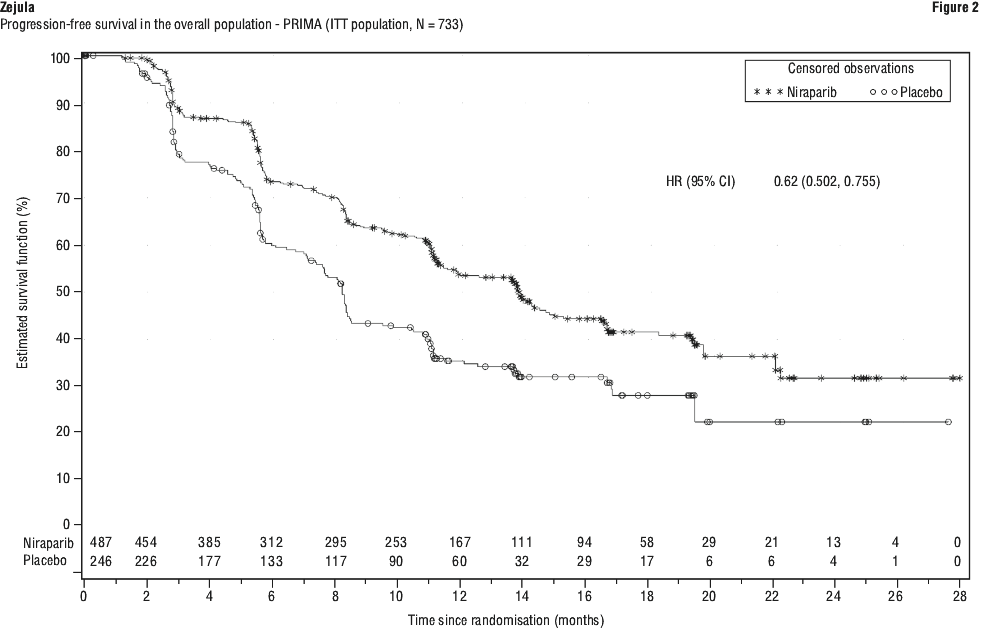
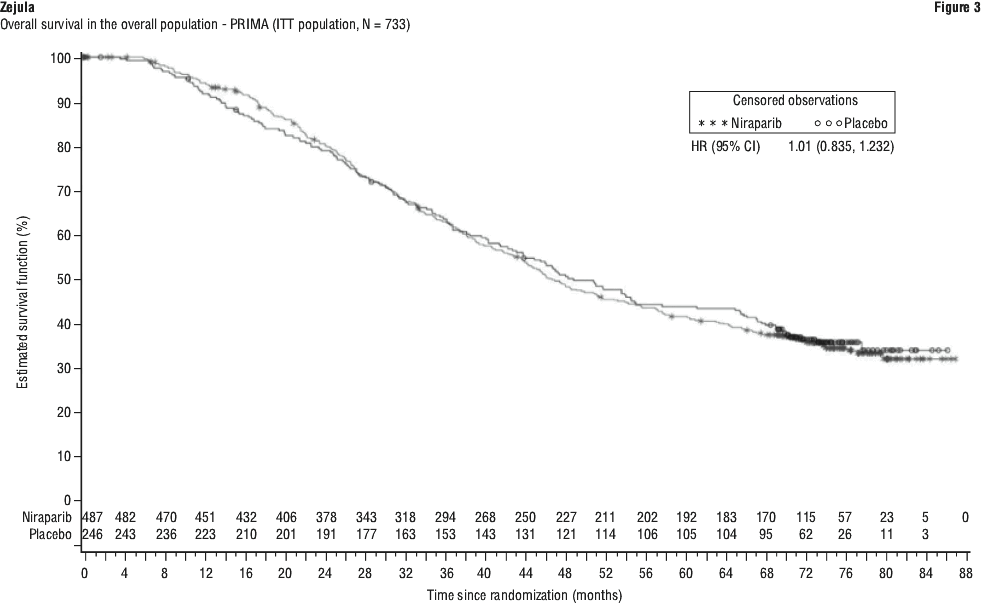
Overall survival analysis in PRIMA. At the final analysis of OS, the median OS in the overall population was 46.6 months (95% CI: 43.7, 52.8) for patients randomised to niraparib compared with 48.8 months (95% CI: 43.1, 61 .0) in the placebo arm, with a hazard ratio of 1 .01 (95% CI: 0.84, 1 .23) (see Figure 3). The maturity of the OS data for the overall population was 62.5%.
The median OS in the HR-deficient population was 71.9 months (95% CI: 55.5, NE) for patients randomised to niraparib compared to 69.8 months (95% CI: 51 .6, NE) in the placebo arm, with a hazard ratio of 0.95 (95% CI: 0.70, 1 .29) (see Figure 4). The maturity of the OS data for the HR-deficient group was 49.6%.
Recurrent ovarian cancer maintenance treatment. The safety and efficacy of niraparib as maintenance therapy was studied in a Phase 3 randomised, double-blind, placebo-controlled international trial (NOVA) in patients with relapsed predominantly high-grade serous epithelial ovarian, fallopian tube, or primary peritoneal cancer who were platinum-sensitive, defined by complete response (CR) or partial response (PR) for more than six months to their penultimate (next to last) platinum-based therapy. To be eligible for niraparib treatment, the patient was required to be in response (CR or PR) following completion of last platinum-based chemotherapy. The CA-125 levels were required to be normal (or a > 90% decrease in CA-125 from baseline) following their last platinum treatment and be stable for at least 7 days. Patients should not have received prior PARP inhibitor (PARPi) therapy, including niraparib. Eligible patients were assigned to one of two cohorts based on the results of a germline BRCA (gBRCA) mutation test. Within each cohort, patients were randomised using a 2:1 allocation of niraparib and placebo. Patients were assigned to the gBRCAmut cohort based on blood samples for gBRCA analysis that were taken prior to randomisation. Testing for gBRCA mutation and HRD was performed using the HRD test on tumour tissue obtained at the time of initial diagnosis or at the time of recurrence.
Randomisation within each cohort was stratified by time to progression after the penultimate platinum therapy before study enrolment (6 to < 12 months and ≥ 12 months); use or not of bevacizumab in conjunction with the penultimate or last platinum regimen; and best response during the most recent platinum regimen (complete response and partial response).
Patients began treatment on Cycle 1/Day 1 (C1/D1) with niraparib 300 mg or matched placebo administered QD in continuous 28-day cycles. Clinic visits occurred each cycle (4 weeks ± 3 days).
In the NOVA study, 48% of patients had a dose interruption in Cycle 1. Approximately 47% of patients restarted at a reduced dose in Cycle 2.
The most commonly used dose in niraparib-treated patients in the NOVA study was 200 mg.
Progression-free survival was determined per RECIST (Response Evaluation Criteria in Solid Tumors, version 1.1) or clinical signs and symptoms and increased CA-125. PFS was measured from the time of randomisation (which occurred up to 8 weeks after completion of the chemotherapy regimen) to disease progression or death.
The primary efficacy analysis for PFS was determined by blinded central independent assessment and was prospectively defined and assessed for the gBRCAmut cohort and the non-gBRCAmut cohort separately.
Demographics, baseline disease characteristics, and prior treatment history were generally well balanced between the niraparib and placebo arms in the gBRCAmut (n = 203) and the non gBRCAmut cohorts (n = 350). Median ages ranged from 57 to 63 years across treatments and cohorts. The primary tumour site in most patients (> 80%) within each cohort was the ovary; most patients (> 84%) had tumours with serous histology. A high proportion of patients in both treatment arms in both cohorts had received 3 or more prior lines of chemotherapy, including 49% and 34% of niraparib patients in the gBRCAmut and non-gBRCAmut cohorts, respectively. Most patients were age 18 to 64 years (78%), Caucasian (86%) and had an ECOG performance status of 0 (68%).
In the gBRCAmut cohort, the median number of treatment cycles was higher in the niraparib arm than the placebo arm (14 and 7 cycles, respectively). More patients in the niraparib group continued treatment for more than 12 months than patients in the placebo group (54.4% and 16.9% respectively).
In the overall non-gBRCAmut cohort, the median number of treatment cycles was higher in the niraparib arm than in the placebo arm (8 and 5 cycles, respectively). More patients in the niraparib group continued treatment for more than 12 months than patients in the placebo group (34.2% and 21.1%, respectively).
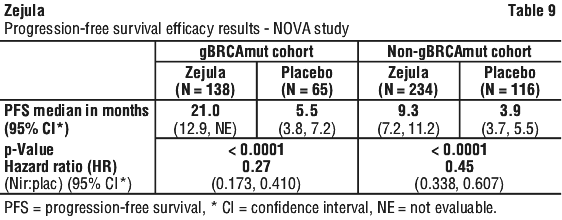
The study met its primary objective of statistically significantly improved PFS for niraparib maintenance monotherapy compared with placebo in the gBRCAmut cohort (HR 0.27; 95% CI* 0.173, 0.410; p < 0.0001) as well as in the overall non-gBRCAmut cohort (HR 0.45; 95% CI* 0.338, 0.607; p < 0.0001). Table 9 shows the results for the PFS primary endpoint for the primary efficacy populations (gBRCAmut cohort and the overall non-gBRCAmut cohort).
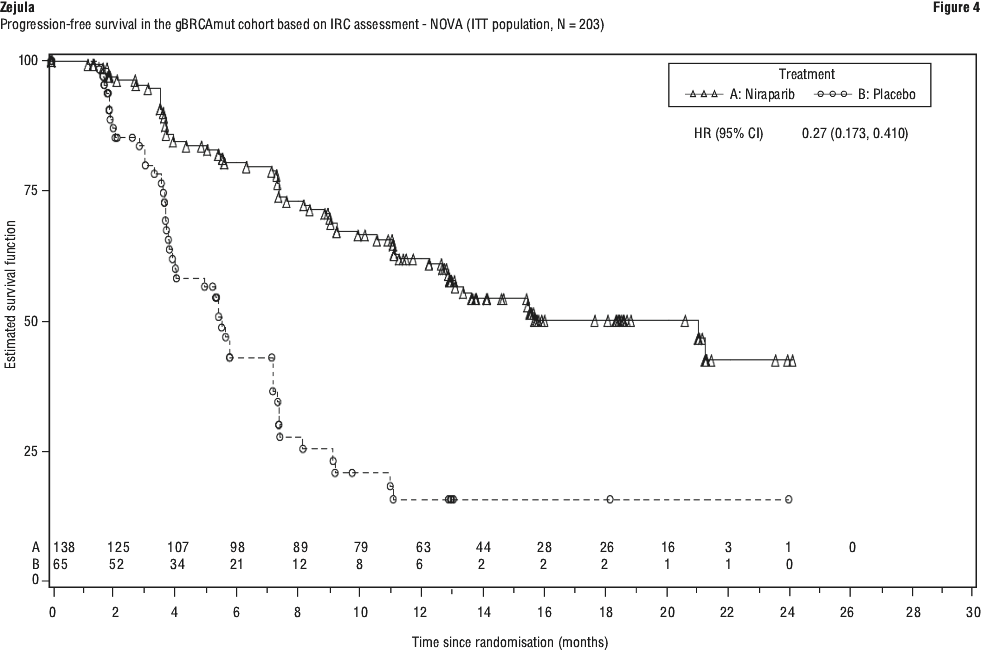
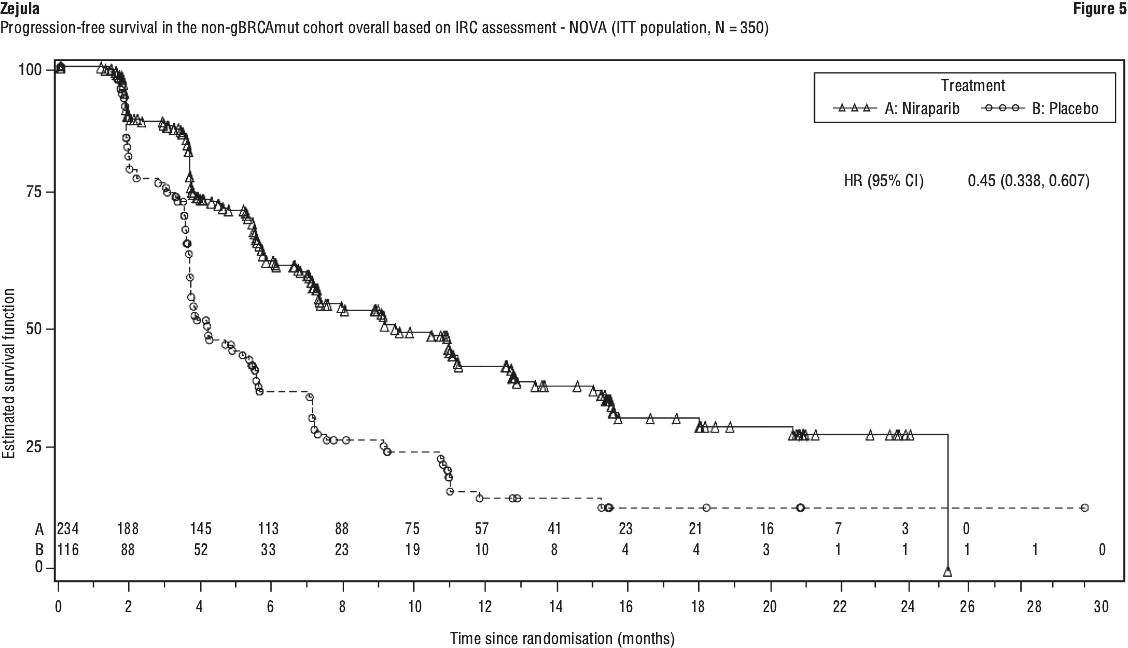
Patient-reported outcomes. Patient-reported outcome (PRO) data from validated survey tools (FOSI and EQ-5D) indicate that niraparib-treated patients reported no difference from placebo in measures associated with quality of life (QoL).
5.2 Pharmacokinetic Properties
Absorption. Following a single-dose administration of 300 mg niraparib under fasting conditions, niraparib was measurable in plasma within 30 minutes and the mean peak plasma concentration (Cmax) for niraparib was reached within 5 hours [range 508 - 875 nanogram/mL across studies]. Following multiple oral doses of niraparib from 30 mg to 400 mg once daily, accumulation of niraparib was approximately 2 to 3-fold.
The systemic exposures (Cmax and AUC) to niraparib increased in a dose-proportional manner when the dose of niraparib increased from 30 mg to 400 mg. The absolute bioavailability of niraparib is approximately 73%, indicating minimal first pass effect.
Administration of niraparib (3 x 100 mg) with a high-fat high-calorie meal may result in a slight decrease in Cmax (~20%) relative to administration of niraparib (3 x 100 mg) under fasted conditions. Food did not significantly affect the overall exposure of niraparib (AUCT and AUC∞).
The tablet and capsule formulations have been demonstrated to be bioequivalent. Following administration of either one 300 mg tablet or three 100 mg capsules of niraparib in 108 patients with solid tumours under fasting conditions, the 90% confidence intervals of the geometric mean ratios for tablet compared to capsules for Cmax, AUClast and AUC∞ fell within the limits of bioequivalence (0.80 and 1.25).
When niraparib tablets were administered following a high-fat meal in patients with solid tumours, niraparib Cmax and AUCinf increased by 11% and 28%, respectively (compared to administration under fasted conditions). Changes of this magnitude are not expected to be clinically meaningful.
Distribution. Niraparib was moderately protein bound in human plasma (83.0%), mainly with serum albumin. In a population pharmacokinetic analysis of niraparib, the Vd/F was 1206 L in cancer patients, indicating extensive tissue distribution of niraparib.
Metabolism. Niraparib is metabolised primarily by carboxylesterases to form a major inactive metabolite, M1. In a mass balance study, M1 and M10 (the subsequently formed M1 glucuronides) were the major circulating metabolites.
Excretion. Following a single oral 300 mg dose of niraparib, the mean terminal half-life (t1/2) of niraparib ranged from 44 to 54 hours (approximately 2 days) across studies. In a population pharmacokinetic analysis, the apparent total clearance (CL/F) of niraparib was 15.9 L/h in cancer patients.
Niraparib is eliminated primarily through the hepatobiliary and renal routes. Following an oral administration of a single 300 mg dose of [14C]-niraparib, on average 86.2% (range 71% to 91%) of the dose was recovered in urine and faeces over 21 days. Radioactive recovery in the urine accounted for 47.5% (range 33.4% to 60.2%) and in the faeces for 38.8% (range 28.3% to 47.0%) of the dose. In pooled samples collected over 6 days, 40.0% of the dose was recovered in the urine primarily as metabolites and 31.6% of the dose was recovered in the faeces primarily as unchanged niraparib.
Special populations. Renal impairment. In the population pharmacokinetic analysis of data from clinical studies in patients, pre-existing mild (ClCr < 90 to ≥ 60 mL/min) and moderate (ClCr < 60 to ≥ 30 mL/min) renal impairment did not influence the clearance of niraparib. No patients with pre-existing severe renal impairment or end stage renal disease undergoing haemodialysis were identified in clinical studies (see Section 4.2 Dose and Method of Administration).
Hepatic impairment. In the population pharmacokinetic analysis of data from clinical studies in patients, pre-existing mild hepatic impairment did not influence the clearance of niraparib.
In a clinical study of cancer patients using NCI-ODWG criteria to classify the degree of hepatic impairment, niraparib AUCinf in patients with moderate hepatic impairment (n = 8) was 1.56 (90% CI: 1.06 to 2.30) times the niraparib AUCinf in patients with normal hepatic function (n = 9) following administration of a single 300 mg dose. Niraparib dose adjustment is recommended for patients with moderate hepatic impairment (see Section 4.2 Dose and Method of Administration). Moderate hepatic impairment did not have an effect on niraparib Cmax or on niraparib protein binding.
The pharmacokinetics of niraparib have not been assessed in patients with severe hepatic impairment (see Section 4.2 Dose and Method of Administration).
Age, weight and race. Population pharmacokinetic analyses indicated that age, weight and race had no significant impact on the pharmacokinetics of niraparib.
Paediatric population. No studies have been conducted to investigate the pharmacokinetics of niraparib in paediatric patients.
5.3 Preclinical Safety Data
Genotoxicity. Niraparib was not mutagenic in a bacterial reverse mutation assay (Ames) test but was clastogenic in an in vitro mammalian chromosomal aberration assay and in an in vivo rat bone marrow micronucleus assay. This clastogenicity is consistent with genomic instability resulting from the primary pharmacology of niraparib and indicates potential for genotoxicity in humans.
Carcinogenicity. Carcinogenicity studies have not been conducted with niraparib.
Animal pharmacology and toxicology. In vitro, niraparib inhibited dopamine (DAT) and norepinephrine (NET) transporters at concentration levels below anticipated human exposure levels (based on unbound Cmax). In mice, single doses of niraparib increased intracellular levels of dopamine and metabolites in the cortex. Reduced locomotor activity was seen in one of two single dose studies in mice. The clinical relevance of these findings is not known but effects on blood pressure and pulse rate that may be related to inhibition of these transporters have occurred in patients.
In repeat-dose oral toxicity studies, niraparib was administered daily for up to 3 months' duration in rats and dogs. The major primary target organ for toxicity in both species was the bone marrow, with associated changes in peripheral haematology parameters. Additionally, decreased spermatogenesis was seen in both species. These findings occurred at exposure levels below those seen clinically, and were largely reversible within 4 weeks of cessation of dosing in dogs but not rats.
6 Pharmaceutical Particulars
6.1 List of Excipients
Capsules. Capsule content. Lactose monohydrate, magnesium stearate.
Capsule shell. Titanium dioxide, gelatin, brilliant blue FCF, erythrosine, tartrazine.
Printing inks. Black Ink; SW-9040 (PI:12418). White Ink; TekPrint SB-0007P White Ink (PI 2216).
Tablets. Tablet core. Microcrystalline cellulose, lactose monohydrate, povidone, crospovidone, silicon dioxide, magnesium stearate.
Film coating. Polyvinyl alcohol, titanium dioxide, macrogol 4000, purified talc, ferrosoferric oxide.
6.2 Incompatibilities
Not applicable.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Capsules. Store below 25°C.
Tablets. Store below 30°C. Store in original container.
6.5 Nature and Contents of Container
Capsules are available in Aclar/PVC/aluminium foil perforated unit dose blisters in cartons of 28, 56, and 84 capsules.
Film-coated tablets are available either in oPA/aluminium/PVC/aluminium/vinyl/acrylic blisters or in oPA/aluminium/PVC/paper layer/aluminium/vinyl/acrylic (child-resistant blisters) in cartons of 84 x 1 and 56 x 1 film-coated tablets.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure. The chemical name for niraparib tosilate monohydrate is (3S)-3-{4-[7-(aminocarbonyl)-2H-indazol-2-yl]phenyl}piperidinium 4-methylbenzenesulfonate monohydrate. The molecular formula is C19H20N4.OC7H8O3S.H2O which corresponds to a formula weight of 510.61 g/mol. Niraparib tosilate monohydrate has one chiral centre of the S configuration. The chemical structure is shown below:
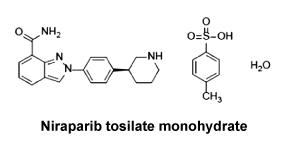
CAS number. 1038915-60-4 (niraparib).
1613220-15-7 (niraparib tosilate monohydrate).
7 Medicine Schedule (Poisons Standard)
Prescription Only Medicine (Schedule 4).
Date of First Approval
28 June 2019
Date of Revision
15 January 2026
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.