Zepzelca
Brand Information
| Brand name | Zepzelca |
| Active ingredient | Lurbinectedin |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Zepzelca.
Summary CMI
ZEPZELCA®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
▼ This medicine is new or being used differently. Please report side effects. See the full CMI for further details.
1. Why am I using ZEPZELCA?
ZEPZELCA contains the active ingredient lurbinectedin. ZEPZELCA has provisional approval in Australia for the treatment of patients with small cell lung cancer, when the cancer has spread to other parts of the body (is metastatic), and has previously been treated with chemotherapy that contained platinum, which either did not work or is no longer working.
For more information, see Section 1. Why am I using ZEPZELCA? in the full CMI.
2. What should I know before I use ZEPZELCA?
Do not use if you have had a severe allergic reaction to lurbinectedin or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use ZEPZELCA? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with ZEPZELCA and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How is ZEPZELCA given?
- The usual dose is 3.2 mg/m2 of body surface area every 3 weeks
- ZEPZELCA will be given to you as an infusion (a drip) into a vein (intravenously) over a period of 60 minutes
More instructions can be found in Section 4. How is ZEPZELCA given? in the full CMI.
5. What should I know while using ZEPZELCA?
| Things you must do |
|
| Things you must not do |
|
For more information, see Section 5. What should I know while using ZEPZELCA? in the full CMI.
6. Are there any side effects?
Common side effects include: anemia, nausea, tiredness and decreased appetite.
Serious side effects that may require urgent medical attention or hospitalisation include: decrease in blood cell counts, serious infections and liver problems.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
▼ This medicine is subject to additional monitoring due to provisional approval. This will allow quick identification of new safety information. You can help by reporting any side effects you may get. You can report side effects to your doctor, or directly at www.tga.gov.au/reporting-problems.
ZEPZELCA®
Active ingredient(s): lurbinectedin
This medicine has provisional registration in Australia for the treatment of patients with metastatic small cell lung cancer (SCLC) that has progressed on or after prior platinum-containing therapy. The decision to provisionally register this medicine has been made on the basis of promising results from preliminary studies. More evidence is required to be submitted when available to substantiate the benefit of the medicine for this use.
Consumer Medicine Information (CMI)
This leaflet provides important information about using ZEPZELCA. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using ZEPZELCA.
Where to find information in this leaflet:
1. Why am I using ZEPZELCA?
2. What should I know before I use ZEPZELCA?
3. What if I am taking other medicines?
4. How is ZEPZELCA given?
5. What should I know while using ZEPZELCA?
6. Are there any side effects?
7. Product details
1. Why am I using ZEPZELCA?
ZEPZELCA contains the active ingredient lurbinectedin. ZEPZELCA is an anti-cancer medicine that works by preventing the tumour cells from multiplying.
ZEPZELCA has provisional approval for the treatment of patients with small cell lung cancer, when the cancer has spread to other parts of the body (is metastatic), and has previously been treated with chemotherapy that contained platinum, which either did not work or is no longer working.
The decision to approve this medicine has been made on the basis of promising results from preliminary studies. More evidence is required to be submitted when available to fully confirm the benefit and safety of the medicine for this use.
It is not known if ZEPZELCA is safe and effective in patients under 18 years old.
2. What should I know before I use ZEPZELCA?
Do not use ZEPZELCA if:
- you are allergic to lurbinectedin, or any of the other ingredients (check the list of ingredients at the end of this leaflet to make sure you can use this medicine).
Before receiving ZEPZELCA, tell your doctor about any medical conditions you have, including if you:
- have problems or abnormal function of your liver or kidney.
- take any other medicines, including over-the-counter medicines, alternative medicines and supplements.
- are pregnant, plan to become pregnant, or are planning to have children in future.
- are breast-feeding.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Female patients who could potentially become pregnant:
- Your doctor may want to do a pregnancy test before you commence treatment with ZEPZELCA.
- You should use highly effective contraception during treatment with ZEPZELCA, and for 7 months after your final dose.
- Tell your doctor immediately if you become pregnant or think you could be pregnant during treatment with ZEPZELCA.
- It is not known if ZEPZELCA passes into breastmilk. You should not breastfeed during treatment with ZEPZELCA.
Male patients with female partners who could potentially become pregnant:
- You should use highly effective contraception during treatment with ZEPZELCA, and for 4 months after your final dose.
Fertility
If you may wish to have children in future, seek medical advice on fertility perseveration treatment prior to therapy with ZEPZELCA, because ZEPZELCA may cause irreversible infertility.
Genetic counselling is also recommended for patients wishing to have children after ZEPZELCA therapy.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interfere with ZEPZELCA and affect how it works, therefore you should avoid the use of ZEPZELCA together with the medicines listed below.
Medicines that may increase the effect of ZEPZELCA include medicines that contain:
- Ketoconazole, itraconazole, posaconazole, voriconazole or fluconazole (for fungal infections),
- ciprofloxacin and erythromycin (for bacterial infections),
- aprepitant (to prevent nausea and vomiting),
- fluvoxamine (for depression and obsessive compulsive disorder [OCD])
- imatinib ( for certain types of leukaemia's)
or - verapamil and diltiazem (for high blood pressure and heart conditions).
Avoid grapefruit products such as grapefruit and grapefruit juice. These products may contain components that increase the effect of ZEPZELCA.
Medicines that may reduce the effect of ZEPZELCA include medicines that contain:
- rifampicin (for bacterial infections),
- phenobarbital, carbamazepine and primidone (for epilepsy)
- bosentan (for high blood pressure in the lungs)
or - St. John's Wort (Hypericum perforatum, an herbal medicine for depression).
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and/or whether they could affect (or be affected by) ZEPZELCA.
4. How is ZEPZELCA given?
ZEPZELCA is given to you in the hospital or clinic under the supervision of a physician experienced in the use of chemotherapy. Its use should be confined to qualified oncologists or other health professionals specialised in the administration of anti-cancer medicines.
How much is given
- The usual dose is 3.2 mg/m2 of body surface area, once every 3 weeks.
- During the treatment period, your doctor will carefully monitor you and decide the most appropriate dosage of ZEPZELCA to give to you.
How it is given
- ZEPZELCA is given as an intravenous (IV) infusion (through a drip into a vein) over a period of 60 minutes.
- ZEPZELCA can be given through a peripheral or central venous line.
- You may be given additional medicines before each treatment (and as needed during the treatment) to help prevent nausea and vomiting, or make it less severe.
How long it will be given for
- The infusion is usually given once every 3 weeks, but your doctor may recommend dose delays if needed to manage side effects.
- The length of your whole treatment period will be determined by your doctor, depending on your progress and how well you feel.
If too much is given (overdose)
As this medicine is being given by your doctor or nurse, it is unlikely that you will be given too much.
In the unlikely event of an overdose, your doctor will monitor you for side effects and treat any symptoms as required.
If you have any further questions on the use of this medicine, ask your doctor.
5. What should I know while using ZEPZELCA?
During treatment with ZEPZELCA, you must inform your doctor or nurse if you feel any of the symptoms mentioned under Section 6. Are there any side effects?. Your doctor might reduce or delay the ZEPZELCA dose, or recommend that you stop treatment, depending on the severity.
Things you must do
- If you are about to be started on any new medicine, remind your doctor that you are being given ZEPZELCA.
- Remind any doctor, dentist or pharmacist you visit that you are using ZEPZELCA.
- You should use highly effective birth control (contraception) during treatment with and for 7 months after your final dose of ZEPZELCA. Males with female partners who are able to become pregnant should use highly effective birth control during treatment with and for 4 months after your final dose of ZEPZELCA.
- Tell your doctor or nurse straight away if you experience an allergic reaction or feel unwell during infusion.
Typical symptoms associated with allergic reactions are redness of the face or chest, itching, coughing, shortness of breath, chest discomfort, etc. Other symptoms may occur as well.
These side effects mostly occur during or after the infusion of the first dose. You will be monitored for signs of these effects during and after the infusion.
Call your doctor straight away if you become pregnant while being given this medicine.
Things you must not do
During your treatment with ZEPZELCA you may feel tired and experience loss of strength. Do not drive or use any tools or machines if you are experiencing any of these side effects.
Storage
ZEPZELCA will be stored in the pharmacy or on the ward. The medicine is kept in its original packaging in the refrigerator where the temperature stays between 2°C and 8°C.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Your doctor will perform certain checks periodically to test for side effects that may affect the blood, liver, kidneys, heart and muscle.
Less serious side effects
| Less serious side effects | What to do |
General or affecting different parts of the body:
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
General or affecting different parts of the body:
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What ZEPZELCA contains
| Active ingredient (main ingredient) | Lurbinectedin |
| Other ingredients (inactive ingredients) | (S)-lactic acid Sucrose Sodium hydroxide Water for Injection |
Do not take this medicine if you are allergic to any of these ingredients.
What ZEPZELCA looks like
ZEPZELCA is a powder for solution for intravenous infusion. The powder has a white to off-white colour. (Aust R 335536).
Who distributes ZEPZELCA
Specialised Therapeutics
Pharma Pty Ltd
Level 2, 17 Cotham Road,
Kew, Victoria, 3101
Ph: 1300 798 820
Fax: 1800 798 829
www.stbiopharma.com
This leaflet was prepared in August 2025.
Brand Information
| Brand name | Zepzelca |
| Active ingredient | Lurbinectedin |
| Schedule | S4 |
▼ This medicinal product is subject to additional monitoring in Australia. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse events at www.tga.gov.au/reporting-problems.
MIMS Revision Date: 01 October 2025
1 Name of Medicine
Zepzelca 4 mg powder for solution for infusion.
2 Qualitative and Quantitative Composition
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
4 mg of lurbinectedin as lyophilised powder in a single-dose vial for reconstitution.
4 Clinical Particulars
4.1 Therapeutic Indications
Zepzelca is indicated for the treatment of patients with metastatic small cell lung cancer (SCLC) that has progressed on or after prior platinum-containing therapy. This indication was approved via the provisional approval pathway, based on objective response rate and duration of response in a single arm trial. Continued approval for this indication depends on verification and description of clinical benefit in a confirmatory trial.
4.2 Dose and Method of Administration
Zepzelca must be administered under the supervision of a physician experienced in the use of chemotherapy. Its use should be confined to qualified oncologists or other health professionals specialised in the administration of cytotoxic agents.
Recommended dose and schedule. The recommended dose is 3.2 mg/m2 by intravenous infusion over 60 minutes, repeated once every 21 days until disease progression or unacceptable toxicity.
Only administer Zepzelca to patients with an absolute neutrophil count above 1.5 x 109/L, and a platelet count above 100 x 109/L.
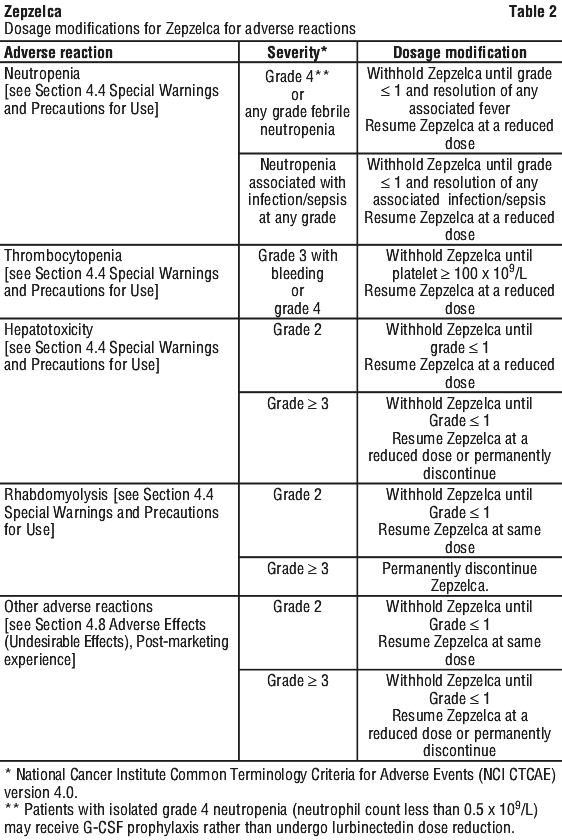
Dose modifications for adverse reactions. The recommended dose reduction levels for adverse reactions are listed in Table 1. Dosage modifications for Zepzelca for adverse reactions are presented in Table 2. Permanently discontinue Zepzelca in patients who are unable to tolerate 2.0 mg/m2 or require a dose delay greater than two weeks.

Corticosteroids (intravenous dexamethasone 8 mg or equivalent);
Serotonin antagonists (intravenous ondansetron 8 mg or equivalent).
Preparation. Zepzelca is a cytotoxic drug. Follow applicable special handling and disposal procedures.
The Zepzelca vial is for single use (in one patient on one occasion) only. Discard any residue.
Prepare the solution for infusion using aseptic technique as follows:
Inject 8 mL of Sterile Water for Injection USP into the vial, yielding a reconstituted solution containing 0.5 mg/mL lurbinectedin.Shake the vial until dissolution is complete.
Visually inspect the reconstituted solution for particulate matter and discoloration. It should be clear, colourless or slightly yellowish, and free of visible particles.
Calculate the required volume of reconstituted solution as follows (see Equation 1):
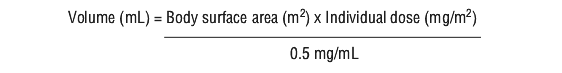
For administration through a peripheral venous line, withdraw the required volume of reconstituted solution from the vial and add to an infusion container containing at least 250 mL of diluent (0.9% Sodium Chloride Injection USP or 5% Glucose Injection).
Use as soon as practicable after reconstitution and dilution. If necessary, the solution can be stored prior to administration, either at room temperature/in ambient light or, ideally, under refrigerated (2° to 8°C) conditions, for a maximum of 24 hours following reconstitution (including infusion time).
Administration. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. If particulate matter is observed, do not administer.
Zepzelca can be administered with or without an in-line filter. If infusion lines containing in-line filters are utilized for administration of Zepzelca, polyethersulfone (PES) in-line filters with pore sizes of 0.22 micron are recommended.
Do not use in-line nylon membrane filters when the reconstituted Zepzelca solution is diluted using 0.9% Sodium Chloride Injection, USP. Adsorption of Zepzelca to the Nylon membrane filters has been observed when 0.9% Sodium Chloride Injection, USP is used as the diluent.
Compatibility with other intravenous administration materials and the diluted Zepzelca solution has been demonstrated in the following materials:
Polyolefin containers (polyethylene, polypropylene and mixtures).
Polyvinyl Chloride (PVC) (non-DEHP-containing), polyurethane and polyolefin infusion sets (polyethylene, polypropylene and polybutadiene).
Implantable venous access systems with titanium and plastic resin ports and with polyurethane or silicone intravenous catheters.
Do not co-administer Zepzelca and other intravenous drugs concurrently within the same intravenous line.
Dose modification for hepatic impairment. Dose adjustment is not required for patients with mild hepatic impairment (total bilirubin ≤ 1.5 x ULN and AST ≤ 3 x ULN). The effect of moderate or severe hepatic impairment (total bilirubin > 1.5 x ULN) on the pharmacokinetics of lurbinectedin is unknown [see Section 5.2 Pharmacokinetic Properties].
Dose modification for renal impairment. Dose adjustment is not required in patients with mild to moderate (CLCR 30-89 mL/min) renal impairment. The effect of severe renal impairment (CLCR < 30 mL/min) or end-stage renal disease on the pharmacokinetics of lurbinectedin is unknown [see Section 5.2 Pharmacokinetic Properties]; therefore, it should not be administered to these patients.
4.3 Contraindications
Zepzelca is contraindicated in patients with history of significant drug allergy to the active substance or any of the excipients.
4.4 Special Warnings and Precautions for Use
Myelosuppression. Zepzelca can cause myelosuppression.
In clinical studies of 554 patients with advanced solid tumours receiving Zepzelca [see Section 4.8 Adverse Effects (Undesirable Effects)], Grade 3 or 4 neutropenia occurred in 41% of patients, with a median time to onset of 15 days and a median duration of 7 days. Febrile neutropenia occurred in 7% of patients. Sepsis occurred in 2% of patients and was fatal in 1% (all cases occurred in patients with solid tumours other than SCLC). Grade 3 or 4 thrombocytopenia occurred in 10%, with a median time to onset of 10 days and a median duration of 7 days. Grade 3 or 4 anaemia occurred in 17% of patients.
Administer Zepzelca only to patients with baseline neutrophil count of at least 1.5 x 109/L and platelet count of at least 100 x 109/L.
Monitor blood counts including neutrophil count and platelet count prior to each administration. For neutrophil count less than 0.5 x 109/L or any value less than lower limit of normal, the use of G-CSF is recommended. Withhold, reduce the dose, or permanently discontinue Zepzelca based on severity [see Section 4.2 Dose and Method of Administration].
Hepatotoxicity. Zepzelca can cause hepatotoxicity.
In clinical studies of 554 patients with advanced solid tumours receiving Zepzelca [see Section 4.8 Adverse Effects (Undesirable Effects)], Grade 3 elevations of ALT and AST were observed in 6% and 3% of patients, respectively, and Grade 4 elevations of ALT and AST were observed in 0.4% and 0.5% of patients, respectively. The median time to onset of Grade ≥ 3 elevation in transaminases was 8 days (range: 3 to 49), with a median duration of 7 days.
Monitor liver function tests prior to initiating Zepzelca and periodically during treatment as clinically indicated. Withhold, reduce the dose, or permanently discontinue Zepzelca based on severity [see Section 4.2 Dose and Method of Administration].
Extravasation resulting in tissue necrosis. Extravasation of Zepzelca resulting in skin and soft tissue injury, including necrosis requiring debridement, may occur. Consider use of a central venous catheter to reduce the risk of extravasation, particularly in patients with limited venous access. Monitor patients for signs and symptoms of extravasation during the Zepzelca infusion. If extravasation occurs, immediately discontinue the infusion, remove the infusion catheter, and monitor for signs and symptoms of tissue necrosis. The time to onset of necrosis after extravasation may vary.
Administer supportive care and consult with an appropriate medical specialist as needed for signs and symptoms of extravasation. Administer subsequent infusions at a site that was not affected by extravasation.
Rhabdomyolysis. Rhabdomyolysis has been reported in patients treated with Zepzelca. Monitor creatine phosphokinase (CPK) prior to initiating Zepzelca and periodically during treatment as clinically indicated. Withhold or reduce the dose based on severity [see Section 4.2 Dose and Method of Administration].
If rhabdomyolysis occurs, supportive measures such as parenteral hydration, urine alkalinization and dialysis should be promptly established, as indicated. Caution should be taken if medicinal products with known association with rhabdomyolysis (e.g. statins), are administered concomitantly with lurbinectedin, since the risk of rhabdomyolysis may be increased.
Use in hepatic impairment. Zepzelca has not been studied in patients with AST > 3 x ULN, or in patients with moderate or severe hepatic impairment (bilirubin > 1.5 x ULN).
Use in renal impairment. Zepzelca has not been studied in patients with severe renal impairment (CLCR < 30 mL/min) or end-stage renal disease.
Embryo-fetal toxicity. Based on animal data and its mechanism of action, Zepzelca can cause fetal harm when administered during pregnancy [see Section 4.6 Fertility, Pregnancy and Lactation].
Test to verify the pregnancy status of females of reproductive potential prior to initiating Zepzelca. Advise patients who are pregnant of the potential risk to the fetus. Advise female patients of reproductive potential to use highly effective contraception during treatment with Zepzelca and for 7 months after the last dose. Advise male patients with female partners of reproductive potential to use highly effective contraception during treatment with Zepzelca and for 4 months after the last dose.
Use in the elderly. Of the 105 patients with SCLC who received Zepzelca in clinical studies, 35% were ≥ 65 years of age, and 9% were ≥ 75 years of age. No overall difference in effectiveness was observed between patients who were ≥ 65 years of age compared to those who were < 65 years of age.
There was a higher incidence of serious adverse reactions in patients who were ≥ 65 years of age compared to those who were < 65 years of age (49% vs. 26%, respectively). The serious adverse reactions most frequently reported in patients ≥ 65 years of age were related to myelosuppression and consisted of febrile neutropenia (11%), neutropenia (11%), thrombocytopenia (8%), and anaemia (8%) [see Section 4.8 Adverse Effects (Undesirable Effects)].
Paediatric use. The safety and effectiveness of Zepzelca in paediatric patients have not been established.
Effects on laboratory tests. See Section 4.8 Adverse Effects (Undesirable Effects).
4.5 Interactions with Other Medicines and Other Forms of Interactions
Strong and moderate CYP3A inhibitors. Coadministration with a strong or a moderate CYP3A inhibitor is expected to increase lurbinectedin systemic exposure [see Section 5.2 Pharmacokinetic Properties] and may increase the incidence and severity of adverse reactions to Zepzelca.
Avoid coadministration of Zepzelca with strong CYP3A inhibitors (e.g. itraconazole, ketoconazole, posaconazole, voriconazole, or grapefruit) or moderate CYP3A inhibitors (e.g. aprepitant, ciprofloxacin, diltiazem, erythromycin, fluconazole, fluvoxamine, imatinib, or verapamil). For moderate CYP3A4 inhibitors, if coadministration with Zepzelca cannot be avoided, consider dose reduction of Zepzelca, based on individual tolerability as clinically indicated [see Section 4.2 Dose and Method of Administration].
Strong and moderate CYP3A inducers. Coadministration with a strong CYP3A inducer is expected to decrease lurbinectedin systemic exposure [see Section 5.2 Pharmacokinetic Properties] and may reduce Zepzelca efficacy. Avoid coadministration of Zepzelca with strong CYP3A inducers (e.g. carbamazepine, phenytoin, rifampicin, or St. John's wort) or moderate CYP3A inducers (e.g. bosentan, phenobarbital, or primidone).
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. Zepzelca can cause embryolethality at doses lower than the human dose of 3.2 mg/m2 [see Section 4.6 Fertility, Pregnancy and Lactation, Use in pregnancy].
No dedicated fertility studies were conducted in animal species. Testicular atrophy and hypospermia were observed in rats and dogs at doses below the recommended clinical dose.
Pregnancy testing. Verify the pregnancy status of females of reproductive potential prior to initiating Zepzelca.
Contraception. Females. Advise female patients of reproductive potential to use highly effective contraception during and for 7 months after the use of Zepzelca.
Males. Advise males with a female sexual partner of reproductive potential to use highly effective contraception during and for 4 months after the use of Zepzelca.
Use in pregnancy. (Category D)
Based on animal data and its mechanism of action [see Section 5.1 Pharmacodynamic Properties], lurbinectedin can cause fetal harm when administered during pregnancy. There are no available clinical data to inform the risk of Zepzelca use during human pregnancy. Intravenous administration of a single lurbinectedin dose (approximately 0.2 times the 3.2 mg/m2 clinical dose) to pregnant rats during the period of organogenesis caused 100% embryolethality.
Advise patients who are pregnant and females of reproductive potential of the potential risk to a fetus.
Use in lactation. There are no data on the presence of lurbinectedin in human milk, its effects on a breastfed child, or its effects on milk production. Because of the potential for serious adverse reactions from Zepzelca in breastfed children, advise patients not to breastfeed during treatment with Zepzelca and for 2 weeks after the final dose.
4.7 Effects on Ability to Drive and Use Machines
No studies on the effects of the ability to drive and to use machines have been performed. However, some events of fatigue, dizziness, vertigo and nausea have been reported in patients receiving lurbinectedin. Patients who experience such events during therapy should not drive or operate machinery.
4.8 Adverse Effects (Undesirable Effects)
The following clinically significant adverse reactions are described in detail in other sections of the prescribing information:
Myelosuppression [see Section 4.4 Special Warnings and Precautions for Use];
Hepatotoxicity [see Section 4.4 Special Warnings and Precautions for Use];
Extravasation resulting in tissue necrosis [see Section 4.4 Special Warnings and Precautions for Use];
Rhabdomyolysis [see Section 4.4 Special Warnings and Precautions for Use].
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in Section 4.4 Special Warnings and Precautions for Use reflects exposure to Zepzelca as a single agent at a dose of 3.2 mg/m2 intravenously every 21 days in 554 patients with advanced solid tumours. Among 554 patients who received Zepzelca, including 105 patients with small cell lung cancer (SCLC) in PM1183-B-005-14 (Study B-005), 24% were exposed for 6 months or longer and 5% were exposed for greater than one year.
Small cell lung cancer (SCLC). The safety of Zepzelca was evaluated in a cohort of 105 patients with previously treated SCLC in Study B-005 [see Section 5.1 Pharmacodynamic Properties, Clinical trials]. Patients received Zepzelca 3.2 mg/m2 intravenously every 21 days. All patients in this study received a pre-specified anti-emetic regimen consisting of a corticosteroid and serotonin antagonist. Patients could receive G-CSF for secondary prophylaxis (i.e. after patients had a first adverse event of leukopenia), but not primary prophylaxis (i.e. prior to any occurrence of leukopenia). Among patients who received Zepzelca, 29% were exposed for 6 months or longer and 6% were exposed for greater than one year.
Serious adverse reactions occurred in 34% of patients who received Zepzelca. Serious adverse reactions in ≥ 3% of patients included pneumonia, febrile neutropenia, neutropenia, respiratory tract infection, anaemia, dyspnoea, and thrombocytopenia.
Permanent discontinuation due to an adverse reaction occurred in two patients (1.9%) who received Zepzelca. Adverse reactions resulting in permanent discontinuation in ≥ 1% of patients who received Zepzelca, which included peripheral neuropathy and myelosuppression.
Dosage interruptions due to an adverse reaction occurred in 30.5% of patients who received Zepzelca. Adverse reactions requiring dosage interruption in ≥ 3% of patients who received Zepzelca included neutropenia, and hypoalbuminaemia.
Dose reductions due to an adverse reaction occurred in 25% of patients who received Zepzelca. Adverse reactions requiring dosage reductions in ≥ 3% of patients who received Zepzelca included neutropenia, febrile neutropenia and fatigue.
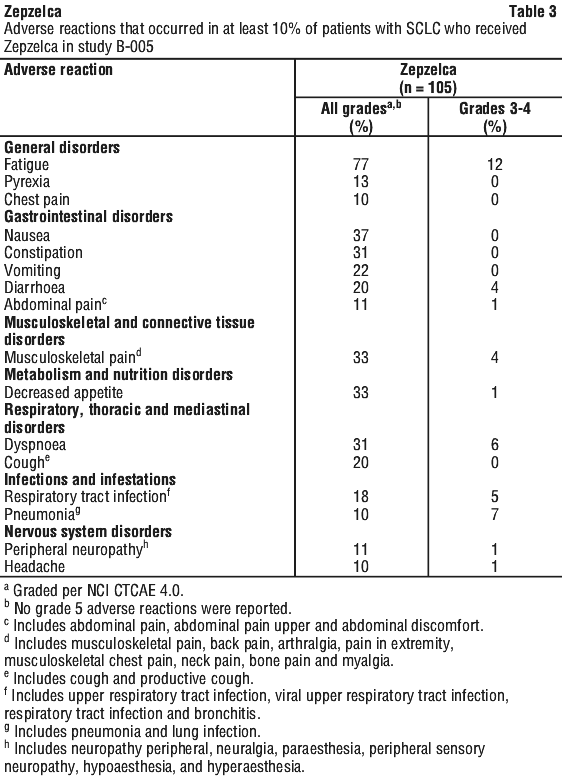
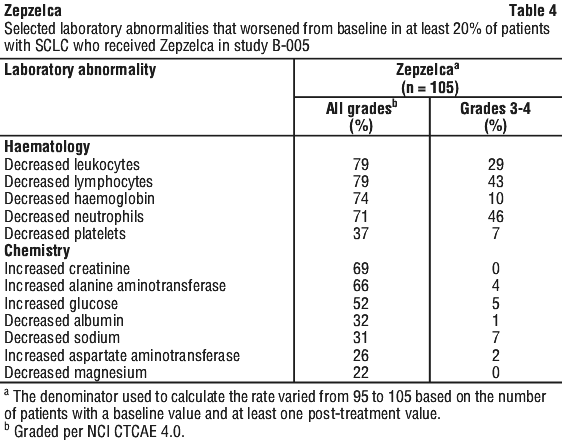
The most common adverse reactions, including laboratory abnormalities, (≥ 20%) were leukopenia, lymphopenia, fatigue, anaemia, neutropenia, increased creatinine, increased alanine aminotransferase, increased glucose, thrombocytopenia, nausea, decreased appetite, musculoskeletal pain, decreased albumin, constipation, dyspnoea, decreased sodium, increased aspartate aminotransferase, vomiting, cough, decreased magnesium and diarrhoea.
The most common adverse reactions, and selected laboratory abnormalities that occurred in the SCLC cohort of Study B-005 are summarised in Table 3 and Table 4, respectively.
General disorders and administration site conditions. Extravasation (including tissue necrosis requiring debridement).
Musculoskeletal and connective tissue disorders. Rhabdomyolysis.
Metabolism and nutrition disorders. Tumour lysis syndrome.
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems and drugsafety-STA@stbiopharma.com.
4.9 Overdose
If an overdose is suspected, monitor the patient closely for myelosuppression and hepatic enzymes and institute supportive care measures as appropriate.
Haemodialysis is not expected to enhance the elimination of Zepzelca because lurbinectedin is highly bound to plasma proteins (99%), and renal excretion is negligible.
There is no known antidote for overdosage with Zepzelca.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action. Lurbinectedin is an alkylating drug that binds guanine residues in the minor groove of DNA, forming adducts and resulting in a bending of the DNA helix towards the major groove. Adduct formation triggers a cascade of events that can affect the subsequent activity of DNA binding proteins, including some transcription factors, and DNA repair pathways, resulting in perturbation of the cell cycle and eventual cell death.
Lurbinectedin inhibited human monocyte activity in vitro and reduced macrophage infiltration in implanted tumours in mice.
Pharmacodynamics. Lurbinectedin exposure-response relationships and the pharmacodynamic time-course for efficacy have not been fully characterised.
Increased incidence of Grade 4 neutropenia and Grade ≥ 3 thrombocytopenia were observed with increased lurbinectedin exposure.
Cardiac electrophysiology. The potential for QT prolongation with lurbinectedin was evaluated in a sub-study of 39 patients with advanced cancer who received lurbinectedin 3.2 mg/m2 every 3 weeks in Study B-005. No large mean increase in QTcF (> 20 ms) was detected. The maximum mean QTcF change from baseline was 5.4 ms (upper bound of 90% CI: 9.6 ms).
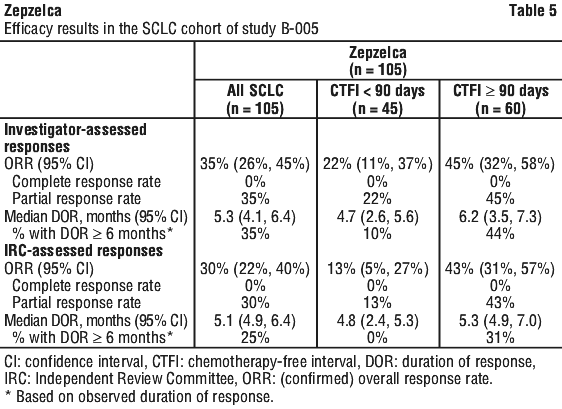
Clinical trials. Study B-005. Study B-005 (PM1183-B-005-14) was an open-label, multicentre, single-arm study evaluating Zepzelca as a single agent in patients with advanced or metastatic solid tumours, including a cohort of patients with small cell lung cancer (SCLC) whose disease had progressed on or after platinum-based chemotherapy. Patients received 3.2 mg/m2 Zepzelca, administered as a 60-minute IV infusion once every 21 days (one cycle). Patients received a median of 4 cycles of Zepzelca (range 1 to 24 cycles). The trial excluded patients with central nervous system (CNS) involvement, grade ≥ 3 dyspnoea, daily intermittent oxygen requirement, hepatitis or cirrhosis, and immunocompromised patients. Tumour assessments were conducted every 6 weeks for the first 18 weeks and every 9 weeks thereafter. The primary efficacy outcome measure was confirmed, investigator-assessed, overall response rate (ORR), using Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. Additional efficacy outcome measures included investigator-assessed duration of response (DOR), as well as ORR and DOR assessed by an Independent Review Committee (IRC).
A total of 105 patients with SCLC who progressed on or after platinum-based chemotherapy were enrolled. The median age was 60 years (range, 40-83 years) and 35% were at least 65 years old. The majority of patients were male (60%) and white (75%); ethnicity was not reported for 23%. Baseline ECOG performance status was 0 or 1 for 92% of patients, and 92% were current/former smokers. All patients had received at least one line of platinum-based chemotherapy for advanced disease (range 1-2 lines), and prior radiotherapy had been administered to 71% of patients. Eight patients (8%) had prior immunotherapy in addition to platinum-based chemotherapy. Sixty patients (57%) had platinum-sensitive SCLC, defined as recurrence or progression ≥ 90 days after the last dose of platinum-containing therapy (chemotherapy free interval [CTFI] ≥ 90 days). The remaining 45 patients had platinum-resistant SCLC, defined as recurrence or progression < 90 days after the last dose of platinum-containing therapy (CTFI < 90 days).
Table 5 summarises investigator-assessed and IRC-assessed key efficacy measures in all patients and in platinum-resistant and platinum-sensitive subgroups.
5.2 Pharmacokinetic Properties
After a 3.2 mg/m2 lurbinectedin dose administered as a 1-hour IV infusion, geometric means (CV%) of total plasma Cmax and AUC0-inf were 107 microgram/L (79%) and 551 microgram.h/L (94%), respectively. No accumulation of lurbinectedin in plasma is observed upon repeated administrations every 3 weeks.
Distribution. The volume of distribution (CV%) of lurbinectedin at steady state is 504 L (39%). Plasma protein binding is approximately 99%, to both albumin and α-1-acid glycoprotein.
Metabolism. Lurbinectedin is metabolised by CYP3A4 in vitro.
Excretion. The terminal half-life of lurbinectedin is 51 hours. Total plasma clearance (CV%) of lurbinectedin is 11 L/h (50%).
After administration of a single dose of radiolabeled lurbinectedin, 89% of the radioactivity was recovered in faeces (< 0.2% unchanged) and 6% was recovered in urine (1% unchanged).
Drug interaction studies. Lurbinectedin is metabolised by CYP3A4 in vitro, and interactions with strong and moderate CYP3A inhibitors and inducers are anticipated. Dedicated clinical drug-drug interaction studies with CYP3A modulators have not been completed.
In a Phase 1 study with lurbinectedin, patients who received aprepitant (a weak-moderate CYP3A4 inhibitor used as an antiemetic) showed a 33% reduction of lurbinectedin plasma clearance when compared with patients who did not receive it.
In vitro data. Cytochrome P450 (CYP) enzymes. Lurbinectedin is metabolised by CYP3A4.
At clinically relevant concentrations, lurbinectedin is not an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4.
Lurbinectedin is not an inducer of CYP1A2 or CYP3A4.
Transporter systems. Lurbinectedin is a substrate of MDR1 (P-gp), but is not a substrate of OATP1B1, OATP1B3, OCT1, or MATE1.
Lurbinectedin inhibits MDR1 (P-gp), OATP1B1, OATP1B3, and OCT1 but not BCRP, BSEP, MATE1, OAT1, OAT3, or OCT2.
Pharmacokinetics in specific populations. No clinically significant differences in the pharmacokinetics of lurbinectedin were identified based on age (range: 18-85 years), sex, body weight (range: 39-154 kg), mild to moderate renal impairment (CLCR 30 to 89 mL/min) or mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN, or total bilirubin between 1.0-1.5 x ULN and any AST). The effects of severe renal impairment (CLcr < 30 mL/min) and moderate or severe hepatic impairment (total bilirubin > 1.5 x ULN and any AST) on the pharmacokinetics of lurbinectedin have not been studied.
5.3 Preclinical Safety Data
Genotoxicity. Lurbinectedin is genotoxic to mammalian cells in the presence and absence of metabolic activation, and was positive in the mouse lymphoma cell assay. Lurbinectedin was not mutagenic in vitro in a bacterial reverse mutation (Ames) assay.
Carcinogenicity. Carcinogenicity testing of lurbinectedin has not been performed.
6 Pharmaceutical Particulars
6.1 List of Excipients
(S)-lactic acid; sucrose; sodium hydroxide.
6.2 Incompatibilities
Zepzelca must not be mixed or diluted with other medicinal products except those mentioned in Section 4.2.
6.3 Shelf Life
Unopened vials. 60 months [see Section 6.4 Special Precautions for Storage].
After reconstitution or dilution. 24 hours, including infusion time. Use immediately if possible [see Section 6.4 Special Precautions for Storage].
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Unopened vials. Refrigerate (2° to 8°C). Refrigerate. Do not freeze.
After reconstitution or dilution. Stable at room temperature with exposure to ambient light for up to 24 hours. However, microbiological hazard is minimized by immediate use, or refrigeration (2° to 8°C) if storage is necessary. Do not store for longer than 24 hours, including infusion time. Do not freeze.
6.5 Nature and Contents of Container
Zepzelca (lurbinectedin) powder for solution for infusion is supplied as a sterile, preservative-free, lyophilised powder in a 30 mL clear glass vial. Each carton contains one single-dose vial.
6.6 Special Precautions for Disposal
Zepzelca is a cytotoxic drug. Follow applicable special handling and disposal procedures.
In Australia, any unused medicine or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
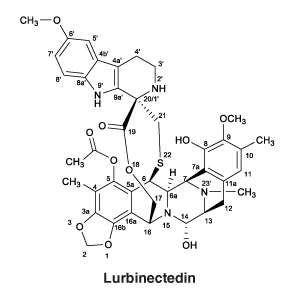
Zepzelca is a synthetic molecule, which binds to the minor groove of DNA and is a selective inhibitor of oncogenic transcription. The chemical name of Zepzelca (lurbinectedin) is (1'R,6R,6aR,7R,13S,14S,16R)-8,14-dihydroxy-6',9-dimethoxy-4,10,23-trimethyl-19-oxo- 2',3',4',6,7,9',12,13,14,16-decahydro-6aH-spiro[7,13-azano-6,16-(epithiopropanooxymethano) [1,3]dioxolo[7,8]isoquinolino[3,2-b][3]benzazocine-20,1'-pyrido[3,4-b]indol]-5-yl acetate.
Molecular formula: C41H44N4O10S.
Molecular weight: 784.87 g/mol.
Chemical structure.
7 Medicine Schedule (Poisons Standard)
Prescription Only Medicine (S4).
Date of First Approval
10 September 2021
Date of Revision
05 September 2025
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.