Hizentra
Brand Information
| Brand name | Hizentra |
| Active ingredient | Immunoglobulin, normal (human) |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Hizentra.
Summary CMI
Hizentra®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I using Hizentra®?
Hizentra® contains the active ingredient human normal immunoglobulin. Hizentra® is used for the replacement of antibodies because your antibody levels are low (referred to as immunodeficiency), or for a condition where there is an imbalance in your immune system requiring treatment with immunoglobulins (referred to as immunomodulation).
For more information, see Section 1. Why am I using Hizentra®? in the full CMI.
2. What should I know before I use Hizentra®?
Do not use if you have ever had an allergic reaction to Hizentra® or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use Hizentra®? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with Hizentra® and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use Hizentra®?
- Your doctor will determine the dose of Hizentra® that you will receive.
- Doses may be given at repeated intervals, from daily to once every two weeks.
Your doctor may adjust the dose based on your response to the treatment. More instructions can be found in Section 4. How do I use Hizentra®? in the full CMI.
5. What should I know while using Hizentra®?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using Hizentra®? in the full CMI.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention. The most common side effects are swelling, pain, redness or itching where the infusion was given. The more serious side effects include reduced urination, severe headache, neck stiffness, inability to stand bright light, painful eye movements, pain/tenderness or swelling/discolouration of an arm or leg, tingling, numbness or weakness on one side of the body, shortness of breath, chest pain, fever or an allergic reaction.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
Hizentra®
Active ingredient: Human normal immunoglobulin
Consumer Medicine Information (CMI)
This leaflet provides important information about using Hizentra®. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using Hizentra®.
Where to find information in this leaflet:
1. Why am I using Hizentra®?
2. What should I know before I use Hizentra®?
3. What if I am taking other medicines?
4. How do I use Hizentra®?
5. What should I know while using Hizentra®?
6. Are there any side effects?
7. Product details
1. Why am I using Hizentra®?
Hizentra® contains the active ingredient human normal immunoglobulin.
Immunoglobulins are also known as antibodies and are blood proteins that help your body to fight infections.
Immunoglobulins are produced by your body's immune system to fight infections caused by bacteria and viruses.
Hizentra® is used:
- for the replacement of antibodies because your antibody levels are low (referred to as immunodeficiency), or
- for a condition where there is an imbalance in your immune system requiring treatment with immunoglobulins (referred to as immunomodulation).
2. What should I know before I use Hizentra®?
Warnings
Do not use Hizentra® if:
- you have a history of allergy to human immunoglobulin products (allergic reactions may include skin rash, face swelling, wheezing or breathing difficulties), or previously been told you react to any of the ingredients listed at the end of this leaflet. Always check the ingredients to make sure you can use this medicine.
- you have been told you have antibodies to immunoglobulin A (IgA)
- you suffer from hyperprolinaemia type I or II (a genetic disorder causing high levels of the amino acid proline in the blood)
Check with your doctor if you:
- are receiving Hizentra® or this type of medicine (a human normal immunoglobulin) for the first time or after a long break in treatment (e.g. several months), or if you have been switched from another human normal immunoglobulin medicine
- have a history of heart, or blood vessel disease, or blood clots, have thick blood, have been immobile for some time. Also tell the doctor what medicine you are using as some medicines, such as those that contain the hormone estrogen (for example, birth control pills), may increase your risk of developing a blood clot.
- have low blood volume (hypovolaemia)
- have IgA deficiency
- have frequent headaches or migraine
- are over 65 years of age
- have any allergies to any medicines, foods, preservatives or dyes.
Vaccinations
Please inform your doctor if you have had any vaccinations within the last two weeks or are planning to have a vaccination. Hizentra® may impair the effect of some virus vaccines such as measles, mumps, rubella and varicella for a period of at least 6 weeks, and up to 3 months. After receiving this medicine, a period of 3 months should be allowed before vaccination with some virus vaccines. In the case of measles vaccine, this effect may last for up to 1 year, so if you are going to receive a measles vaccine you should have your measles antibody status checked.
Tell your vaccinating doctor about your treatment with Hizentra® before receiving any vaccination.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
Virus safety
Hizentra® is made from human plasma (the liquid component of blood). When medicines are made from human blood or plasma, processes are used to prevent infections being passed from the blood/plasma donor to the person receiving the medicine.
These processes include careful selection of the people who donate blood and plasma to make sure that those who might be carrying infections are excluded. In addition, each donation and pools of donations are tested for indicators of virus/virus infection(s).
Manufacturers of these medicines also include steps in the processing of blood or plasma that inactivate or remove viruses. Despite these processes, when medicines are prepared from human blood or plasma, the possibility of passing on an infection cannot be totally ruled out. Unknown or new viruses or other types of infection could also be passed on.
However, the measures taken in the manufacture of this medicine are considered effective for enveloped viruses such as human immunodeficiency virus (HIV), hepatitis B virus, and hepatitis C virus, and for the non-enveloped viruses hepatitis A (HAV) and B19 virus (B19V).
There is reassuring clinical experience regarding the lack of HAV or B19V infections with immunoglobulins. It is assumed that the antibodies which are in the immunoglobulin product make an important contribution to the viral safety.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interfere with Hizentra® and affect how it works.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect Hizentra®.
4. How do I use Hizentra®?
How much to use
- Treatment should be started and supervised by a doctor. Hizentra® is administered as an infusion subcutaneously (under the skin). If your doctor decides that you should receive Hizentra® at home, they will ensure you receive detailed instructions and training on how to use it.
- Your doctor will determine the dose of Hizentra® that you will receive. Doses may be given at repeated intervals, from daily to once every two weeks. Your doctor may adjust the dose based on your response to the treatment.
When to use Hizentra®
- Your doctor will discuss with you when Hizentra® should be used.
How to prepare Hizentra® for administration
If your doctor considers that you should receive Hizentra® at home, the instructions below should be followed carefully.
Step 1: Clean surface
Clean a table or other flat surface.
Step 2: Assemble supplies
Gather the Hizentra® vial(s) or pre-filled syringe(s) and the following supplies (not provided with Hizentra®), as directed by your doctor. The Hizentra® vials/pre-filled syringes must be at room temperature before administration.
- Administration tubing
- Subcutaneous needle and required tubing
- Syringes
- Transfer device or drawing up needle(s)
- Gauze and tape, or transparent dressing (if required)
- Infusion pump (if required)
- Sharps container
- Alcohol (antiseptic) wipes
- Treatment diary.
Step 3: Wash hands
Thoroughly wash and dry your hands (Figure 1).

Step 4: Check vials or pre-filled syringes
If using pre-filled syringes,
carefully peel back the transparent covering from the tray and inspect the protective cap. Peel back the outer layer of the wrap-around label to allow for viewing of Hizentra® through the fully transparent inner layer, but don't remove the label completely (Figure 2).

If using vials, inspect the protective cap of the vials (Figure 3).
Hizentra® is a pale-yellow to light-brown clear solution. Check for particles or colour changes.

Do not use the pre-filled syringe or vial if:
- the liquid looks cloudy, contains particles, or has changed colour.
- the protective cap of the pre-filled syringe or the vial is missing or defective.
- the expiry date on the label has passed.
Step 5: Preparation of Hizentra® for infusion
- If using Hizentra® pre-filled syringes, go to Step 5.1
- If using Hizentra® vials, go to Step 5.2
Step 5.1: Hizentra® pre-filled syringe(s)
The 5 mL and 10 mL pre-filled syringes are fully assembled and ready for use (Figure 4).

For the 20 mL and 50 mL pre-filled syringe, screw the plunger rod onto the pre-filled syringe stopper prior to use (Figure 5).

If you are using a syringe pump, Hizentra® pre-filled syringes can be placed directly in the syringe pump if the syringe size matches the pump requirements. Please follow the manufacturer's instructions.
If the pre-filled syringe does not match the infusion pump requirements, transfer the contents of the pre-filled syringe to another syringe of a size specific for the infusion pump by following the directions below:
Use a syringe-to-syringe transfer device (tip-to-tip connector). (Figure 6)

Remove the protective cap from the pre-filled syringe. Attach the transfer device by twisting it onto the pre-filled syringe. Attach the empty syringe by screwing it onto the other side of the transfer device (Figure 7).

Push the plunger of the pre-filled syringe to transfer Hizentra® from the pre-filled syringe to the empty syringe.
- Repeat this step if multiple pre-filled syringes are necessary to achieve the prescribed dose. Remove the empty pre-filled syringe and attach another pre-filled syringe to the transfer device.
After the transfer is complete, remove the empty pre-filled syringe and the transfer device by unscrewing them from the syringe specific for your pump. Connect the filled syringe to the infusion tubing.
Go to Step 6.
Step 5.2: Transfer Hizentra® from vial(s) to syringe
Take the protective cap off the vial (Figure 8).

Clean the vial stopper with an alcohol wipe (Figure 9). Allow to dry.

Attach a needle or transfer device to the syringe tip, using a clean (non-touch) technique. If using a transfer device, follow the instructions provided by the device manufacturer. If using a needle and a syringe to transfer Hizentra®, follow the instructions below.
- Attach a sterile transfer needle to a sterile syringe (Figure 10).

- Pull out the plunger of the syringe to fill the syringe with air. Make sure the amount of air is the same as the amount of Hizentra® you will transfer from the vial.
- Put the Hizentra® vial on a flat surface. Keeping the vial upright, insert the transfer needle into the centre of the rubber stopper.
- Check that the tip of the needle is not in the liquid. Then, push the plunger of the syringe down. This will inject the air from the syringe into the airspace of the vial.
- Leaving the needle in the stopper while keeping pressure on the syringe plunger, carefully turn the vial upside down (Figure 11).

- Gently withdraw the needle so the tip sits in the Hizentra® fluid and release the pressure on the plunger. The fluid will automatically fill the syringe. Once the automatic filling is complete slowly pull back on the plunger to fill the syringe with any remaining Hizentra®.
- Take the filled syringe and needle out of the stopper. Take off the needle and throw it away in the sharps container.
- When using multiple vials to achieve the desired dose, repeat this step.
Step 6: Prepare the Subcutaneous needles and related tubing
Attach the subcutaneous needle to infusion tubing (if required).
Prime (fill) the infusion tubing. To prime the tubing, connect the syringe filled with Hizentra® to the infusion tubing and gently push on the syringe plunger to fill the tubing with Hizentra® (Figure 12).

Stop priming before Hizentra® fluid reaches the needle.
Step 7: Prepare infusion site(s)
Select an area on your abdomen, thigh, upper arm, or side of upper leg/hip area for the infusion (Figure 13).

Never infuse into areas where the skin is tender, bruised, red, or hard. Avoid infusing into scars or stretch marks.
The number of infusion sites depends on the volume of the total dose. Your healthcare professional can advise you about this.
If you are using more than one infusion site, be sure each site is at least 5 cm apart.
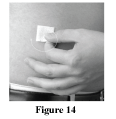
Clean the skin at each site with an alcohol wipe (Figure 14). Let the skin dry.
Step 8: Insert needle(s)
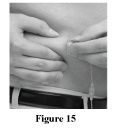
Using two fingers, pinch the skin around the infusion site. Insert the needle into the skin (Figure 15). Your healthcare professional will advise you on the positioning of the needle as it is dependent on the type of needle used.
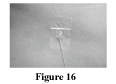
Secure the needle in place as directed by your healthcare professional (Figure 16). This will keep the needle from coming out
Step 9: Start infusion
MAKE SURE YOU ARE NOT INFUSING Hizentra® INTO A BLOOD VESSEL. To test for this, pull the plunger back gently. If you see any blood flowing back into the tubing, remove the needle. Discard the tubing and needle and restart from Step 6.
If using an infusion pump, prepare the pump following the manufacturer's instructions.
Follow the instructions that you have been given by your healthcare professional to start the infusion. Infuse at the rate you have been instructed (Figure 17).

Step 10: Record treatment
Complete your treatment diary as instructed by your healthcare professional.
Step 11: Clean up
Remove the needle set and cover the infusion site with a protective dressing as directed by your healthcare professional.
Discard the empty Hizentra® pre-filled syringe(s) or vial(s), needles, infusion tubing, and syringes as directed by your healthcare professional (Figure 18).
General rubbish such as packaging can be discarded in the general waste.
If using an infusion pump, clean and store it, following the manufacturer's instructions.
If you forget to use Hizentra®
If you think you have missed a dose, speak to your doctor as soon as possible.
If you use too much Hizentra®
The effects of an overdose of Hizentra® are not known.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using Hizentra®?
Things you should do
Tell your doctor if you are planning to travel overseas.
It is important to obtain a written statement from your doctor explaining the reasons why you need to have this medicine and infusion devices with you, otherwise you may not be allowed to bring it into the country of travel.
Please ensure you have multiple copies of the letter if travelling to more than one country.
Call your doctor straight away if you:
- become pregnant while using this medicine.
- are about to have any blood test. Hizentra® may interfere with the results of some tests, resulting in misleading results for some things.
Remind any doctor, dentist or pharmacist you visit that you are using Hizentra®.
Things you should not do
- Do not change the dose or dosing interval without consulting with your doctor. If you think you should receive Hizentra® more or less frequently, please speak to your doctor.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how Hizentra® affects you.
Hizentra® is not expected to affect your ability to drive a car or operate machinery.
Looking after your medicine
- Store below 25°C. Do not freeze.
- Keep the vial or pre-filled syringe in the outer carton in order to protect from light.
Keep it where young children cannot reach it.
When to discard your medicine
Hizentra® does not contain any preservatives, so any unused portion should be discarded immediately.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What Hizentra® contains
| Active ingredient (main ingredient) | Human normal immunoglobulin |
| Other ingredients (inactive ingredients) | Proline Polysorbate 80 Water |
Do not take this medicine if you are allergic to any of these ingredients.
What Hizentra® looks like
Hizentra® is a clear and colourless or pale-yellow to light-brown solution.
Hizentra® is packaged using latex free materials.
Vials:
1 g in a 5 mL solution: AUST R 207386
2 g in a 10 mL solution: AUST R 207385
4 g in a 20 mL solution: AUST R 207383
10 g in a 50 mL solution: AUST R 207384
Pre-filled syringes:
1 g in a 5 mL solution: AUST R 285344
2 g in a 10 mL solution: AUST R 285345
4 g in a 20 mL solution: AUST R 329342
10 g in a 50 mL solution: AUST R 457854
Who distributes Hizentra®
Hizentra® is supplied in Australia by
CSL Behring (Australia) Pty Ltd
ABN 48 160 734 761
189–209 Camp Road
Broadmeadows VIC 3047
Australia
For Medical/Technical Enquiries
TOLL FREE: 1800 642 865
For Customer Service Enquiries
TOLL FREE: 1800 063 892
customerservice@cslbehring.com.au
www.cslbehring.com.au
This leaflet was prepared in May 2025.
® Registered trademark of CSL Limited Group of Companies
Brand Information
| Brand name | Hizentra |
| Active ingredient | Immunoglobulin, normal (human) |
| Schedule | S4 |
MIMS Revision Date: 01 April 2025
1 Name of Medicine
Normal immunoglobulin (human) 20% (20 g/100 mL), subcutaneous injection.
2 Qualitative and Quantitative Composition
Hizentra is a 20% solution containing 20 g/100 mL of total human plasma protein with a purity of at least 98% immunoglobulin G (IgG). More than 90% of the IgG consists of monomers and dimers, aggregates (≤ 2%, typically below 0.1%). The distribution of the IgG subclasses is similar to that of normal human plasma (approximate values: 69% IgG1, 26% IgG2, 3% IgG3, 2% IgG4).
The maximum IgA content is 0.05 mg/mL (normally below 0.005 mg/mL).
The product contains 250 mmol/L of proline as a stabiliser which is a physiological nonessential amino acid. The product also contains trace amounts of polysorbate 80 and sodium. Hizentra contains no carbohydrate stabiliser (e.g. sucrose, maltose) and no preservative.
3 Pharmaceutical Form
Hizentra is a sterile, clear and colourless or pale-yellow or light-brown solution of human normal immunoglobulin for subcutaneous injection.
Hizentra has a nominal osmolality of 380 mOsm/kg and is approximately isotonic. The pH value of the ready-to-use solution is 4.8.
4 Clinical Particulars
4.1 Therapeutic Indications
Replacement therapy in adults and children in:
primary immunodeficiency disease (PID); and
symptomatic hypogammaglobulinaemia secondary to underlying disease or treatment.
Immunomodulatory therapy in:
Chronic Inflammatory Demyelinating Polyneuropathy (CIDP) as maintenance therapy after stabilisation with IVIg.
4.2 Dose and Method of Administration
Hizentra should only be administered subcutaneously.
Dosage. The dose and dose regimen are dependent on the indication.
Replacement therapy (PID and symptomatic hypogammaglobulinaemia). The dose may need to be individualised for each patient dependent on the clinical response and serum IgG trough levels. The following dose regimens are given as a guideline.
The dose regimen using the subcutaneous route should achieve a sustained level of IgG. A loading dose of at least 0.2 to 0.5 g/kg (1.0 to 2.5 mL/kg) bodyweight (bw) may be required. This may need to be divided over several days. After steady-state IgG levels have been attained, maintenance doses are administered at repeated intervals, from daily to once every two weeks, to reach a cumulative monthly dose of the order of 0.4 to 0.8 g/kg (2.0 to 4.0 mL/kg) bw. Trough levels should be measured and assessed in conjunction with the patient's clinical response. Depending on the clinical response (e.g. infection rate), adjustment of the dose and/or the dose interval may be considered in order to aim for higher trough levels.
Paediatric population (PID). As the dose is given by body weight and adjusted to the clinical outcome of the above mentioned conditions the dosage regimen is the same in the paediatric population as in adults. See Section 4.4 Special Warnings and Precautions for Use.
Immunomodulatory therapy (CIDP). The therapy with Hizentra is initiated 1 week after the last IVIg infusion. The recommended subcutaneous dose is 0.2 to 0.4 g/kg bw per week. The weekly dose can be divided into smaller doses and administered by the desired number of times per week. For dosing every two weeks, double the weekly Hizentra dose.
The dose may need to be adapted to achieve the desired clinical response. The patient's individual clinical response should be the primary consideration in dose adjustment.
Hizentra was not evaluated in clinical studies in patients with CIDP who are under the age of 18.
Elderly population. The dose in the elderly population is not considered to be different from that in patients 18 to 65 years of age as the dose is given by body weight and adjusted to the clinical outcome. See Section 4.4 Special Warnings and Precautions for Use.
Administration. This product is normally clear and pale-yellow or light-brown. If it appears to be cloudy or contains particulate matter, do not use product but return the unopened vial or prefilled syringe to CSL Behring.
Hizentra should only be administered subcutaneously. Do not administer intravenously.
Hizentra should be at room temperature before use. Do not shake the Hizentra vial or prefilled syringe.
Hizentra may be infused subcutaneously into sites such as abdomen, thigh, upper arm, and/or lateral hip. If large doses are given (> 50 mL), it may be advisable to administer the dose at multiple sites. There is no limit to the number of infusion sites administered in parallel. Two infusion devices can be used simultaneously. The volume of product infused into a particular site may vary. Infusion sites should be at least 5 cm apart.
Hizentra can be infused using: an infusion device or by manual push with a syringe.
The recommended infusion rate depends on the individual patient's needs:
Device-assisted infusion. The initial infusion rate should not exceed 20 mL/hour/site.
If well-tolerated (also see Section 4.4 Special Warnings and Precautions for Use), the infusion rate can then gradually be increased to 35 mL/hour/site for the subsequent two infusions. Thereafter, the infusion rate can be further increased as per patient's individual tolerability.
Manual push infusion. The recommended initial infusion rate should not exceed 0.5 mL/min (30 mL/hour/site).
If well-tolerated, the infusion rate can be increased up to 2.0 mL/min/site (120 mL/hour/site), based on the healthcare professional's judgement and patient's individual tolerability. See Table 1.

Only one infusion site per syringe can be infused at a time. Do not reuse the needle.
Data on higher infusion volumes per site and higher infusion rates in manual push and pump-assisted administration techniques are limited (see Section 4.8 Adverse Effects (Undesirable Effects)).
Home treatment. If the supervising physician believes that home administration is appropriate, the patient or caregiver must be instructed in: subcutaneous administration techniques; the keeping of a treatment diary; recognition of and measures to be taken in case of severe adverse reactions.
4.3 Contraindications
Hizentra is contraindicated in patients with a history of severe systemic hypersensitivity or anaphylactic reactions/anaphylaxis to the active substance of Hizentra or to any of its excipients.
Hizentra is contraindicated in patients with hyperprolinaemia type I or II.
4.4 Special Warnings and Precautions for Use
Hizentra is for subcutaneous use only. If Hizentra is accidentally administered into a blood vessel, patients could develop shock.
The recommended infusion rate given under Administration should be adhered to. Patients should be closely monitored and carefully observed for any adverse events throughout the infusion period.
Hypersensitivity. Hypersensitivity reactions may occur even in patients who had tolerated previous treatment with human normal immunoglobulin. Severe hypersensitivity or anaphylactic reactions up to shock can particularly occur in patients with known allergies to anti-IgA antibodies. Patients with anti-IgA antibodies, in whom treatment with subcutaneous IgG products remains the only option, should be switched to Hizentra only under close medical supervision.
In case of severe hypersensitivity/anaphylactic reactions the administration of Hizentra must be stopped immediately. In case of shock, standard medical treatment should be administered.
Potential complications can often be avoided by ensuring that patients:
are not sensitive to human normal immunoglobulin, by initially injecting the product slowly;
are carefully monitored for any symptoms throughout the infusion period. In particular, patients naive to human normal immunoglobulin, patients switched from an alternative product or when there has been a long interval since the previous infusion should be monitored during the first infusion and for the first hour after the first infusion, in order to detect potential adverse signs. All other patients should be observed for at least 20 minutes after administration.
Embolic and thrombotic events. Arterial and venous thromboembolic events have been associated with the use of immunoglobulins. Caution should be taken in patients with pre-existing risk factors for thromboembolic events, such as advanced age, estrogen use, in-dwelling vascular catheters, a history of vascular disease or thrombotic episodes, cardiovascular risk factors (including history of atherosclerosis and/or impaired cardiac output), patients with acquired or inherited hypercoagulable states, patients with prolonged periods of immobilisation, severely hypovolaemic patients, patients with diseases which increase blood viscosity (including cryoglobulins, fasting chylomicronaemia and/or high triglyceride levels, and monoclonal gammopathies). In patients at risk for thromboembolic adverse reactions, Hizentra should be administered subcutaneously at the minimum rate of infusion and dose practicable, and these individuals should be monitored for thrombotic complications. Consideration should also be given to measurement of baseline blood viscosity in individuals at risk for hyperviscosity.
Patients should be sufficiently hydrated before use of immunoglobulins.
Aseptic meningitis syndrome (AMS). AMS has been reported with use of IVIg or SCIg. The syndrome usually begins within several hours to 2 days following IgG treatment. AMS is characterised by the following signs and symptoms: severe headache, neck stiffness, drowsiness, fever, photophobia, nausea, and vomiting. Patients exhibiting signs and symptoms of AMS should receive a thorough neurological examination, including cerebrospinal fluid studies, to rule out other causes of meningitis. Discontinuation of IgG treatment may result in remission of AMS within several days without sequelae.
Pathogen safety. Hizentra is made from human plasma. Standard measures to prevent infections resulting from the use of medicinal products prepared from human blood or plasma include selection of donors, screening of individual donations and plasma pools for specific markers of infection and the inclusion of effective manufacturing steps for the inactivation/removal of viruses.
Despite these, when medicinal products prepared from human blood or plasma are administered, the possibility of transmitting infective agents cannot be totally excluded. This also applies to unknown or emerging viruses and other pathogens.
The measures taken are considered effective for enveloped viruses such as HIV, HBV and HCV, and for the nonenveloped viruses HAV and parvovirus B19.
There is reassuring clinical experience regarding the lack of hepatitis A or parvovirus B19 transmission with Hizentra and it is also assumed that the antibody content makes an important contribution to the viral safety.
It is strongly recommended that every time Hizentra is administered to a patient, the name and batch number of Hizentra are recorded in order to maintain a link between the patient and the batch of the product.
Use in the elderly. Clinical studies in PID included 9 patients over the age of 65. Of the 172 CIDP patients evaluated in the PATH study, 34 subjects treated with Hizentra were > 65 years of age. In these studies, there were no elderly-specific dose requirements necessary to achieve the desired serum IgG levels.
Paediatric use. Hizentra was evaluated in 62 paediatric patients (33 children [2 to < 12 years] and 29 adolescents [12 to < 18 years]) with primary immunodeficiency disease (PID). No paediatric-specific dose requirements were necessary to achieve the desired serum IgG levels.
Clinical trials with Hizentra showed a similar safety profile in paediatric and adult patients with PID. The safety and efficacy of Hizentra has not been formally studied in paediatric patients under two years of age.
Hizentra was not evaluated in clinical studies in patients with CIDP who are under the age of 18.
Effects on laboratory tests. Interference with serological testing. After infusion of immunoglobulin the transitory rise of the various passively transferred antibodies in the patient's blood may result in misleading positive results in serological testing.
Passive transmission of antibodies to erythrocyte antigens, e.g. A, B and D may interfere with some serological tests for red cell allo-antibodies (Coombs' test).
4.5 Interactions with Other Medicines and Other Forms of Interactions
Live attenuated virus vaccines. Immunoglobulin administration may impair for a period of at least 6 weeks and up to 3 months the efficacy of live attenuated virus vaccines such as measles, rubella, mumps and varicella. After administration of this medicinal product, an interval of 3 months should elapse before vaccination with live attenuated virus vaccines. In the case of measles, this impairment may persist for up to 1 year. Therefore patients receiving measles vaccine should have their antibody status checked. Refer to the Australian Immunisation Handbook for clinical practice recommendations.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. Animal fertility studies have not been conducted with Hizentra. Based on clinical experience with immunoglobulins it is suggested that no harmful effects on fertility are to be expected.
Use in pregnancy. Animal reproduction studies have not been conducted with Hizentra. Data from prospective clinical trials on the use of human normal immunoglobulin in pregnant women is limited. Therefore, Hizentra should only be given with caution to pregnant women and breast-feeding mothers. Clinical experience with immunoglobulins suggests that no harmful effects on the course of pregnancy, or on the foetus or the neonate are to be expected.
There was no evidence of a teratogenic effect of the excipient proline when administered to pregnant rats over the period of organogenesis.
Continued treatment of the pregnant woman ensures a passive immunity for the neonate.
Use in lactation. During breast-feeding immunoglobulins are excreted into the milk and may contribute to the transfer of protective antibodies to the neonate.
4.7 Effects on Ability to Drive and Use Machines
Hizentra has no or negligible influence on the ability to drive and use machines.
4.8 Adverse Effects (Undesirable Effects)
Experience from clinical studies. In view of the fact that clinical trials are conducted under controlled conditions, adverse reaction (AR) rates observed in the clinical trials of a drug product may not reflect the rates observed in clinical practice.
Tabulated summary of adverse reactions. The Adverse Effects (AEs) have been collected in Hizentra clinical trials from 7 phase III studies in patients with PID (n = 231), 2 phase IV studies in patients with PID (n = 74), 1 phase III study (n = 115) and 1 extension study (n = 82) in patients with CIDP (total N = 502).
The AEs reported in these clinical studies have been assessed. Those considered adverse reactions are summarised and categorised according to the MedDRA System Organ Class (SOC) and Preferred Term (PT) level. The frequency per patient and per infusion has been evaluated using the following criteria: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1000 to < 1/100), rare (≥ 1/10,000 to < 1/1000), very rare (< 1/10,000). See Table 2.
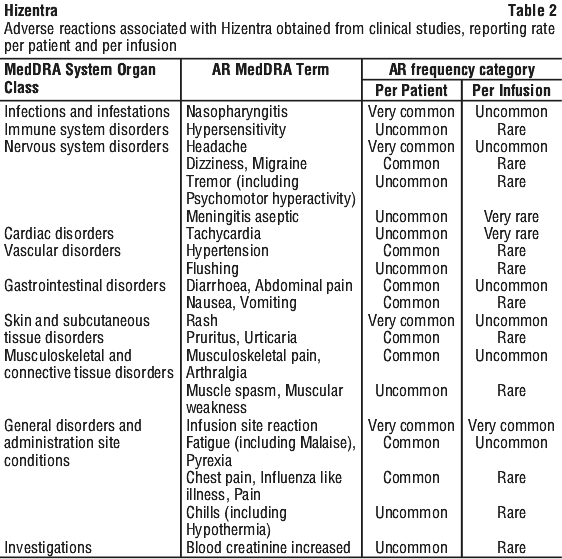
Immune system disorders. Anaphylactic reaction.
Nervous system disorders. Burning sensation.
Vascular disorders. Embolic and thrombotic events.
General disorders and administration site conditions. Infusion site ulcer.
Reliable estimates of the frequency of these reactions or establishment of a causal relationship to product exposure are not possible because the reporting is voluntary and from a population of uncertain size.
Safety profile of higher infusion volumes per site and higher infusion rates in manual push and pump-assisted administration techniques. In the study assessing the safety and tolerability of higher infusion volumes per site and higher infusion rates applied via the manual push and pump-assisted administration, a total of 49 PID patients were enrolled into the corresponding 3 groups (see Section 5.1 Pharmacodynamic Properties). There were no clinically relevant differences in the frequency, type, or intensity of adverse events among the 3 groups. The most frequent adverse events were local site reactions of mild or moderate intensity. The number, type, intensity, or duration of local site reactions did not increase with an increasing infusion parameter in any of the study cohorts. No relevant differences were observed between the BMI ≥/< 30 kg/m2 (n = 14) and age (>/< 18 years) sub-groups. Pump infusion rates up to 100 mL/hour/site, infusion volumes up to 50 mL/site and manual push administration with flow rates up to 2.0 mL/min/site (120 mL/hour/site) were well-tolerated. The safety profile was similar to that of previous PID studies.
Class effects. Infusion site reactions for SCIg.
Paediatric population. Clinical trials with Hizentra showed a similar safety profile in paediatric and adult patients with PID. The safety and efficacy of Hizentra has not been formally studied in paediatric patients under two years of age.
Hizentra was not evaluated in clinical studies in paediatric patients with CIDP who are under the age of 18.
Elderly population. Information available from clinical trials showed no difference in safety profile in patients ≥ 65 years of age than in younger patients. Postmarketing experience with Hizentra in patients ≥ 65 years of age shows an overall similar safety profile in this age group as in younger patients.
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
4.9 Overdose
Consequences of an overdose are not known. In case of overdosing the occurrence of adverse drug reactions should be closely monitored and, if necessary, supporting measures should be offered.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Hizentra contains functionally intact IgG with a broad spectrum of antibodies against infectious agents. It has a distribution of IgG subclasses closely proportional to that in native human plasma. The Fc and Fab functions of the IgG molecule are retained.
Hizentra contains the IgG antibodies present in the normal population. It is prepared from plasma from 1000 or more human donors. The manufacturing process for Hizentra includes three steps to reduce the possibility of virus transmission. Two of these are dedicated virus clearance steps: pH 4 incubation to inactivate enveloped viruses and nanofiltration to remove, by size exclusion, both enveloped and nonenveloped viruses as small as approximately 20 nanometres. In addition, a depth filtration step contributes to the virus reduction capacity.
Mechanism of action. Adequate doses of this medicine may restore abnormally low IgG levels to the normal range.
The mechanism of action in CIDP is not fully understood, but may include immunomodulatory effects.
Clinical trials. The safety and efficacy of Hizentra has been assessed in 7 phase III studies and 2 phase IV studies in patients with PID, and in 1 phase III study including 1 extension study in patients with CIDP.
Primary immunodeficiency diseases (PID). In the prospective, open-label, multicentre, single-arm European study, a total of 51 subjects aged between 3 and 60 years were enrolled from previous treatment with intravenous immunoglobulin (IVIg) (60.9%) or subcutaneous immunoglobulin (SCIg) (39.1%) and switched to Hizentra. The objectives of the study were to investigate the efficacy, tolerability, safety, pharmacokinetics, and health-related quality of life (HRQL) of Hizentra in subjects with PID.
Hizentra was administered subcutaneously for up to 41 weeks, at a dose equivalent to the previous Ig dose. The mean dose administered each week was 0.12 g/kg bw, over a median infusion duration up to 1.25 hours during the wash-in/wash-out period; and up to 1.27 hours during the efficacy period (weeks 13 to 40). Subjects received in total 1831 weekly Hizentra infusions.
The primary endpoint was to demonstrate in the intention-to-treat population (n = 46) sustained total serum IgG trough values, comparable to those observed with the previous IgG treatment. The mean of individual median IgG trough values increased by 8.1% with Hizentra treatment (from 7.49 g/L with the previous IgG therapy to 8.10 g/L during infusions 12 to 17), thus meeting the primary endpoint.
No serious bacterial infections (SBIs) were reported during the efficacy period. The annual rate of total infections/ subject/ year was 5.18 (95% confidence limits: 4.305; 6.171). Twenty subjects in the ITT population (43.5%) missed work/ school/ kindergarten/ day care or were unable to perform normal activities due to infections for 198 days, equating to an annual rate of 8.00 days/ subject/ year. Four subjects (8.7%) were hospitalised due to infections during the efficacy period for a total of 86 days, with an annual rate of 3.48 days/ subject/ year. Thirty two subjects (69.6%) were treated with antibiotics on 1743 days, with an annual rate of 72.75 days/ subject/ year.
Some aspects of HRQL and treatment satisfaction improved with Hizentra treatment compared to previous IVIg treatment, with statistically significant improvements from baseline observed for the treatment satisfaction questionnaire for medication (TSQM) domain convenience and for the total life quality index score. No differences were observed for Hizentra compared to previous SCIg treatment.
In the US a prospective, open-label, multicentre, single-arm, clinical study evaluated the efficacy, tolerability, and safety of Hizentra in 49 adult and paediatric PID subjects, 2 to 75 years of age. Subjects previously receiving monthly treatment with IVIg were switched to weekly subcutaneous administration of Hizentra for 15 months (a 3-month wash-in/wash-out period followed by a 12-month efficacy period). The efficacy analyses included 38 subjects in the modified intention-to-treat population. The primary endpoint was annual rate of SBIs per subject.
A dose adjustment coefficient was applied when switching from IVIg to Hizentra to ensure comparable systemic IgG exposure. Weekly doses of Hizentra ranged from 0.07 to 0.38 g/kg which was 149% (range: 114% to 180%) of the previous IVIg dose. Subjects received a total of 2264 infusions. The number of injection sites per infusion ranged from 1 to 12, with ≤ 4 used in 73% of infusions.
The annual SBI rate per subject was zero (upper 99% confidence limit: 0.132), thus achieving the primary efficacy endpoint.
Sustained IgG trough levels with a mean concentration of 12.53 g/L were achieved throughout the treatment period. The annual rate of any infections was 2.76 infections/ subject/ year (95% CI: 2.235; 3.370), in 31 subjects (81.6%). Twelve subjects (31.6%) missed work/ school/ kindergarten/ day care or were unable to perform normal activities on 71 days, with an annual rate of 2.06 days/ subject/ year. One subject was hospitalised seven days due to infections, equating to an annual rate of 0.20 days/ subject/ year. Twenty-seven subjects (71.1%) were treated with antibiotics on 1688 days, with an annual rate of 48.5 days/ subject/ year.
The Japanese multicentre, single-arm, prospective, open-label, Phase III study evaluated the efficacy, safety, tolerability, HRQL, pharmacoeconomics and PK of Hizentra in subjects with PID.
Twenty-five adult and paediatric subjects who were receiving regular IVIg treatment received 3 prospective IVIg infusions, followed by weekly subcutaneous Hizentra infusions for up to 24 weeks, at doses equal to their previous IVIg weekly equivalent dose. Primary efficacy analysis was based on 21 patients in the per protocol group.
Patients received in total 584 weekly infusions. The mean dose administered in the efficacy period was 87.81 mg/kg bw. Sustained IgG trough levels with mean IgG concentrations of 7.21-7.53 g/L were achieved throughout the efficacy period. There was an increase in the mean of individual median IgG trough values of 9% following switch to Hizentra, from 6.53 g/L in the IVIg period to 7.15 g/L, with a geometric mean ratio of 1.09. No patient had a serious bacterial infection during any part of the study. The annualised rate of infections, hospitalisations, days missed from school or work and antibiotic use were similar for the IVIg and SCIg periods.
To assess the safety and tolerability of higher infusion rates applied via the manual push and pump-assisted administration technique, 49 PID subjects aged 2 to 75 years were enrolled in an open-label, multicentre, parallel-arm, nonrandomised phase IV study and treated with Hizentra for at least 12 weeks (11 paediatric patients aged 2 to < 18, 35 adult patients aged 18 to 65, and 3 geriatric patients aged > 65 years). The study used descriptive statistics and differences between groups were not tested for statistical significance. In the patient group (n = 16) receiving Hizentra via the manual push technique (manual push flow rate cohort), 2 to 7 infusions per week were administered with the flow rates of 25-30, 60 and 120 mL/hour/site corresponding to 0.5 mL/min, 1.0 mL/min or 2.0 mL/min per site (30, 60 or 120 mL/hour/site). Out of 16 subjects in this cohort, 8 subjects (50%) administered Hizentra twice a week. The other 8 subjects administered Hizentra 5 times a week (4 subjects), 3 times a week (2 subjects), 6 and 7 times a week (1 subject each). In the patient group (n = 18) receiving Hizentra via pump-assisted administration (pump-assisted flow rate cohort), weekly Hizentra infusions were administered with 25, 50, 75 and 100 mL/hour/site flow rate. In addition, higher infusion volumes of 25, 40 and 50 mL per site (pump-assisted volume cohort) were evaluated in pump-assisted administration of weekly Hizentra doses (n = 15). In all three groups, each infusion parameter was used for 4 weeks, after which tolerating subjects could switch to the next higher infusion parameter.
Overall, the tolerability* was ≥ 0.98 for all infusion parameter levels in all cohorts. The percentage of subjects responding to a higher infusion parameter (= responder**) was: in the manual push flow rate cohort 100.0% at the 30 mL/hour and 60 mL/hour, and 87.5% at the 120 mL/hour per site; in the pump-assisted flow rate cohort 77.8% at the 25 mL/hour and the 50 mL/hour, 66.7% at the 75 mL/hour, and 61.1% at the 100 mL/hour per site; in the pump-assisted volume cohort 86.7% at the 25 mL and 73.3% at the 40 mL and 50 mL per site. No clinically relevant differences in the serum IgG trough concentrations were observed between the baseline at day 1 and at the end of the study in all subjects.
*Tolerability: number of infusions without severe local reactions divided by the total number of infusions.
**Responder: Pump-assisted cohorts: a subject who performed ≥ 3 valid infusions out of 4 for an infusion parameter.
Manual push flow rate cohort: a subject who performed the minimum number of valid infusions (≥ 60%) for an infusion parameter level.
An infusion rate was considered valid, if ≥ 95% of the planned flow rate/volume per ≥ 1 infusion site was achieved.
Chronic inflammatory demyelinating polyneuropathy (CIDP). The safety, efficacy and tolerability of Hizentra in patients with CIDP has been assessed in a multicentre, double-blind, randomised, placebo-controlled, parallel-group phase III PATH (Polyneuropathy and Treatment with Hizentra) study. 172 subjects previously treated with IVIg were randomised to weekly 0.2 g/kg Hizentra, weekly 0.4 g/kg Hizentra or placebo groups, and followed for a subsequent 24 weeks. The mean duration of exposure was 118.9 days in the 0.2 g/kg and 129 days in the 0.4 g/kg Hizentra group (maximum exposure up to 167 and 166 days in each group, respectively). Subjects generally used 4 infusion sites in parallel (up to 8 sites in parallel). In total, 57 subjects received 1514 infusions in the placebo group, 57 subjects received 2007 infusions in the 0.2 g/kg Hizentra group, and 58 subjects received 2218 infusions in the 0.4 g/kg Hizentra group.
The primary efficacy endpoint was the percentage of subjects who had a CIDP relapse or were withdrawn for any other reason in the Hizentra treatment period. CIDP relapse was defined as an increase (deterioration) by at least 1 point in the adjusted Inflammatory Neuropathy Cause and Treatment (INCAT) score compared with baseline excluding (i) an increase in INCAT score of 1 point if this is only due to an increase of the arm score from 0 to 1 (not clinically meaningful worsening) or (ii) an unchanged adjusted INCAT score compared with baseline, where the arm score decreased from 1 to 0 and the leg score increased by 1 point (not clinically meaningful improvement or worsening).
Both Hizentra doses demonstrated superiority over placebo for the primary endpoint. A statistically significant lower percentage of subjects treated with Hizentra, 32.8% for 0.4 g/kg and 38.6% for 0.2 g/kg, had CIDP relapse or was withdrawn for other reasons compared with 63.2% subjects treated with placebo (p < 0.001 or p = 0.007, respectively). When only considering relapse, the CIDP relapse rates were 19.0% for 0.4 g/kg Hizentra and 33.3% for 0.2 g/kg Hizentra compared with 56.1% for placebo (p < 0.001 or p = 0.012, respectively). Accordingly, over the treatment period for up to 24 weeks Hizentra prevented relapse in 81% and 67% of subjects in the 0.4 g/kg and 0.2 g/kg group, respectively, while in the placebo group 44% of subjects remained relapse-free.
Time to CIDP relapse was evaluated, and the corresponding probabilities for CIDP relapse based on Kaplan-Meier estimates on completion were: placebo, 58.8%; 0.2 g/kg Hizentra, 35.0%; and 0.4 g/kg Hizentra, 22.4%. The hazard ratios (95% CI) for the lower dose and higher dose compared to placebo was 0.48 (0.27, 0.85) and 0.25 (0.12, 0.49), respectively.
In the efficacy scores (INCAT score, mean grip strength, and Medical Research Council (MRC) sum score), subjects in both Hizentra dose groups remained stable while subjects in the placebo group deteriorated. Subjects in the high dose Hizentra group remained stable in the Rasch-built Overall Disability Scale (R-ODS) centile score.
A phase III, multicentre, 48-week open-label extension study enrolled 82 CIDP patients from the PATH study. The extension study investigated the long-term safety and efficacy of Hizentra maintenance therapy in the 0.2 g/kg and 0.4 g/kg bw weekly doses.
Due to the design change after commencement of the extension study (affecting 63 patients), some patients switched dosing from 0.2 g/kg bw to 0.4 g/kg bw, or vice versa during the study. For this reason 72/82 subjects received doses of 0.4 g/kg and 73/82 subjects received doses of 0.2 g/kg during the safety and efficacy evaluation period. The mean efficacy evaluation period for the extension study was 125.8 days (range: 1-330) in the 0.2 g/kg, and 196.1 days (range: 1-330) in the 0.4 g/kg bw group.
Of the 18 patients who completed the pivotal PATH study without relapse on 0.4 g/kg bw dose and received 0.4 g/kg bw from enrolment for up to 24 weeks in the extension study one relapsed (5.6%).
Of the 6 patients who completed the PATH study without relapse on 0.2 g/kg bw dose and received this dose in the extension study, 3 patients relapsed, however, the patient cohort was small. Down-titrating patients in the extension study (who were stable and not on placebo in the pivotal study) from 0.4 g/kg to 0.2 g/kg bw dose without occurrence of relapse was possible in 67.9% of subjects (19/28 patients); all relapsers recovered within 4 weeks after treatment with 0.4 g/kg bw dose.
Compared with the baseline measurement in the extension study, grip strength, MRC sum score, and R-ODS centile score remained stable for patients who never had a relapse in the 48-week extension study period.
5.2 Pharmacokinetic Properties
Absorption and distribution. Following subcutaneous administration of Hizentra, peak serum levels are achieved after approximately 2 to 3 days.
Elimination. IgG and IgG-complexes are broken down in cells of the reticuloendothelial system.
Primary immunodeficiency disease (PID). In a clinical phase III trial with Hizentra (n = 46), the subjects achieved sustained trough levels (median 8.1 g/L) over a period of 28 (± 1) weeks when receiving median weekly doses of 0.06 to 0.24 g/kg bw.
The pharmacokinetics were evaluated in a subset of 23 subjects with primary immunodeficiency (PID) (see Table 3).

Simulation of 2-3 missed daily doses resulted in a median serum IgG level decrease of ≤ 4% compared to consistent daily dosing. By replacing the missed doses when daily dosing was resumed, the median concentration profile recovered within 2 to 3 days. However, if missed doses were not replaced when dosing was resumed, it took up to 5-6 weeks for the IgG trough levels to return to steady state.
In a phase IV study evaluating higher Hizentra infusion parameters in manual push and pump-assisted administration, serum IgG trough levels were measured in 49 PID subjects at the end of study versus baseline at day 1 after enrolment. No clinically relevant differences in the serum IgG trough concentrations were observed between the baseline at day 1 and at the end of the study.
Paediatric population. No differences were seen in the pharmacokinetic parameters between adult and paediatric PID study patients.
Chronic inflammatory demyelinating polyneuropathy (CIDP). In the Polyneuropathy and Treatment with Hizentra (PATH) study, subjects (n = 172) achieved sustained trough levels over a period of 24 weeks when receiving weekly doses of 0.2 g/kg and 0.4 g/kg, respectively. The mean (SD) IgG trough concentration after 24 weeks of Hizentra treatment in the 0.4 g/kg group was 20.6 (3.24) g/L and 15.4 (3.06) g/L in the 0.2 g/kg group. Simulations with population-pharmacokinetic models in the PATH study suggest that a comparable IgG exposure (Cmax, AUC0-14 days, Cmin, 14 days) is achieved when the double weekly Hizentra dose is administered every two weeks in the CIDP subjects. These simulations further suggest that a comparable IgG exposure is correspondingly achieved when the weekly maintenance dose of Hizentra is divided in several, more frequent doses (2 to 7 times per week) in the CIDP patient population.
5.3 Preclinical Safety Data
Genotoxicity. No genotoxicity studies have been conducted with Hizentra. The excipient proline was not genotoxic in a standard array of genotoxicity tests.
Carcinogenicity. No carcinogenicity studies have been conducted with Hizentra.
6 Pharmaceutical Particulars
6.1 List of Excipients
See Section 2 Qualitative and Quantitative Composition.
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the ARTG. The expiry date can be found on the packaging.
Use in one patient on one occasion only. Hizentra should be administered as soon as possible after opening the vial/ prefilled syringe as the solution contains no preservative.
6.4 Special Precautions for Storage
Do not use after the expiry date printed on the carton and label. Store below 25°C (do not freeze).
Keep the vial or the prefilled syringe in the outer carton in order to protect from light.
6.5 Nature and Contents of Container
Hizentra is provided as a 20% (20 g/100 mL) ready-to-use solution for subcutaneous administration in single-use vials or single-use prefilled syringes.
5, 10 or 20 mL of solution in a vial (type I glass), 50 mL of solution in a vial (type II glass), with a stopper (halobutyl), a cap (aluminium crimp) and a flip off disc (plastic).
5, 10, 20 or 50 mL of solution in a prefilled syringe (cyclo-olefin-copolymer (COC)).
The product is supplied in the following pack sizes:
One vial of 5 mL solution containing 1 g human normal immunoglobulin.
One vial of 10 mL solution containing 2 g human normal immunoglobulin.
One vial of 20 mL solution containing 4 g human normal immunoglobulin.
One vial of 50 mL solution containing 10 g human normal immunoglobulin.
One prefilled syringe of 5 mL solution containing 1 g human normal immunoglobulin.
One prefilled syringe of 10 mL solution containing 2 g human normal immunoglobulin.
One pre-filled syringe of 20 mL solution containing 4 g human normal immunoglobulin.
One pre-filled syringe of 50 mL solution containing 10 g human normal immunoglobulin.
Note that not all presentations may be available.
6.6 Special Precautions for Disposal
Any unused solution should be discarded appropriately.
6.7 Physicochemical Properties
CAS number. 9007-83-4.
7 Medicine Schedule (Poisons Standard)
S4.
Date of First Approval
08 May 2014
Date of Revision
17 February 2025
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.