Iressa
Brand Information
| Brand name | Iressa |
| Active ingredient | Gefitinib |
| Schedule | S4 |
Consumer Medicine Information (CMI) leaflet
Please read this leaflet carefully before you start using the Iressa.
Summary CMI
IRESSA®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about taking this medicine, speak to your doctor or pharmacist.
1. Why am I taking IRESSA?
IRESSA contains the active ingredient gefitinib. IRESSA is used to treat non-small cell lung cancer, which is one type of lung cancer.
For more information, see Section 1. Why am I taking IRESSA? in the full CMI.
2. What should I know before I take IRESSA?
Do not take it if you have ever had an allergic reaction to IRESSA or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I take IRESSA? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with IRESSA and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I take IRESSA?
- The usual adult dose is one 250 mg tablet taken each day.
- Swallow your IRESSA tablet whole with a glass of water. Do not chew or crush the tablets.
More instructions can be found in Section 4. How do I take IRESSA? in the full CMI.
5. What should I know while taking IRESSA?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while taking IRESSA? in the full CMI.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, some can be minor and temporary. However, some side effects may be serious and possibly fatal, therefore will need immediate medical attention. See Section 6 in the full CMI (6. Are there any side effects?) and, if you need to, ask your doctor if you have any further questions about side effects. Tell your doctor if you experience any side effects, including those not listed in this leaflet.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
Full CMI
IRESSA®
Active ingredient(s): gefitinib
Consumer Medicine Information (CMI)
This leaflet provides important information about taking IRESSA. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about taking IRESSA.
Where to find information in this leaflet:
1. Why am I taking IRESSA?
2. What should I know before I take IRESSA?
3. What if I am taking other medicines?
4. How do I take IRESSA?
5. What should I know while taking IRESSA?
6. Are there any side effects?
7. Product details
1. Why am I taking IRESSA?
IRESSA contains the active ingredient gefitinib. IRESSA belongs to a group of medicines called antineoplastics. These medicines work by stopping cancer cells from growing and multiplying.
IRESSA is used to treat non-small cell lung cancer, which is one type of lung cancer.
Your doctor will have explained why you are being treated with IRESSA and told you what dose to take.
Your doctor may prescribe this medicine for another use. Ask your doctor if you want more information.
This medicine is available only with a doctor's prescription.
IRESSA is not addictive.
2. What should I know before I take IRESSA?
Warnings
Do not take IRESSA if:
- you are allergic to gefitinib, or any of the ingredients listed at the end of this leaflet.
- Always check the ingredients to make sure you can take this medicine.
Symptoms of an allergic reaction to IRESSA may include:
- shortness of breath, wheezing, difficulty breathing or a tight feeling in your chest
- swelling of the face, lips, tongue or other parts of the body
- rash, itching, hives or flushed, red skin
- dizziness or light-headedness
- back pain
Check with your doctor if you:
- have any other medical conditions:
- eye problems. You may need further examination.
- lung problems
- liver problems - take any medicines for any other condition
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
Do not take IRESSA if you are pregnant.
It is not known if it is safe for you to take IRESSA while you are pregnant. It may affect your baby if you take it at any time during pregnancy.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
Do not breastfeed while taking IRESSA.
IRESSA passes into breast milk and therefore there is a possibility that the breast-fed baby may be affected.
Children
Do not give IRESSA to children.
There is no experience of its use in children.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Tell your doctor if you are taking any other medicines, including:
- itraconazole & ketoconazole (medicines used to treat fungal infections)
- metoprolol (beta-blocker)
- rifampicin (antibiotic)
- warfarin (blood thinner)
- phenytoin, carbamazepine, barbiturates, St John's Wort
- vinorelbine (cancer drug)
- medicines that alter the acidity
of the stomach e.g. antacids, H2 antagonists and proton pump inhibitors
These medicines may interfere with IRESSA and affect how it works.
You may need different amounts of your medicine, or you may need to take/use/have different medicines. Your doctor or pharmacist will advise you.
Your doctor and pharmacist may have more information on medicines to be careful with or avoid while taking IRESSA.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect IRESSA.
4. How do I take IRESSA?
Follow all directions given to you by your doctor or pharmacist carefully.
They may differ from the information contained in this leaflet.
Your doctor or pharmacist will tell you how many tablets you will need to take each day.
If you do not understand the instructions on the box, ask your doctor or pharmacist for help.
How much to take
- The usual adult dose is one 250 mg tablet taken each day.
Swallow your IRESSA tablet whole with a glass of water. Do not chew or crush the tablets.
For patients with swallowing difficulties the tablet can be dissolved in drinking water (non-carbonated). No other liquids should be used. Follow these instructions carefully:
- Drop the tablet into half a glass of drinking water (non-carbonated). DO NOT CRUSH THE TABLET.
- Stir the water until the tablet dissolves and the water becomes a cloudy pale orange solution (approximately 10 minutes).
- Drink the liquid immediately.
- Rinse the empty glass with half a glass of drinking water and drink the water.
This cloudy pale orange solution can also be given to patients via a nasogastric tube.
- Follow the instructions provided and take IRESSA until your doctor tells you to stop.
When to take IRESSA
- IRESSA should be taken regularly at about the same time each day. Taking IRESSA at the same time each day will help you remember to take it.
- It does not matter if you take IRESSA with or without food.
- Continue taking IRESSA for as long as your doctor tells you.
When to take IRESSA
- Continue taking IRESSA for as long as your doctor tells you.
If you forget to take IRESSA
IRESSA should be taken regularly at the same time each day. If you miss your dose at the usual time, take it as soon as you remember, as long as it is 12 hours before the next dose is due.
If it is less than 12 hours to the next dose do not take the dose you have missed.
Do not take a double dose to make up for the dose you missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much IRESSA
If you think that you have taken too much IRESSA you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
Symptoms of an IRESSA overdose include the side effects listed below Section 6. Are there any side effects, but are usually of a more severe nature.
5. What should I know while taking IRESSA?
Things you should do
Be sure to keep all your appointments with your doctor so your progress can be checked.
Your doctor may want to do some tests from time to time to check your progress and investigate any unwanted side effects.
Call your doctor straight away if you:
- become pregnant
- experience any of the serious side effects listed in Section 6. Are there any side effects.
Remind any doctor, dentist or pharmacist you visit that you are taking IRESSA.
If you are about to be started on any new medicine, tell your doctor, dentist or pharmacist that you are taking IRESSA.
If you go into hospital, please let the medical staff know you are taking IRESSA.
Things you should not do
- Do not stop taking this medicine suddenly.
- Do not stop taking IRESSA without checking with your doctor
- Do not give IRESSA to anyone else, even if they have the same condition as you.
- Do not take IRESSA if the packaging is torn or shows signs of tampering.
- Do not take IRESSA to treat any other complaints unless your doctor tells you to.
Driving or taking machines
Be careful before you drive or use any machines or tools until you know how IRESSA affects you.
Some patients may feel weak.
Looking after your medicine
- Keep your IRESSA tablets in the blister foil until it is time to take them. If you take IRESSA out of the blister foil, it will not keep well.
- Keep it in a cool dry place where the temperature stays below 30°C.
Follow the instructions in the carton on how to take care of your medicine properly.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Heat and dampness can destroy some medicines.
Keep it where young children cannot reach it.
A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Getting rid of any unwanted medicine
If you no longer need to take this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not take this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
IRESSA may be associated with changes in your blood, urine or liver. Your doctor may want to perform tests from time to time to check on your progress and detect any unwanted side effects.
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What IRESSA contains
| Active ingredient (main ingredient) | gefitinib |
| Other ingredients (inactive ingredients) |
|
Do not take this medicine if you are allergic to any of these ingredients.
What IRESSA looks like
IRESSA 250 mg tablets are brown, round, film-coated tablets. One side of the tablet is marked "IRESSA 250".
IRESSA tablets are packed in blister foils of 30 tablets.
(AUST R 90010)
Who distributes IRESSA
AstraZeneca Pty Ltd
ABN 54 009 682 311
66 Talavera Road
MACQUARIE PARK NSW 2113
Telephone:- 1800 805 342
This leaflet was prepared on December 2025
IRESSA is a trademark of the AstraZeneca group of companies.
© AstraZeneca, 2025
VV-RIM-01413203 v5.0
Brand Information
| Brand name | Iressa |
| Active ingredient | Gefitinib |
| Schedule | S4 |
MIMS Revision Date: 01 March 2021
1 Name of Medicine
Gefitinib.
2 Qualitative and Quantitative Composition
Each tablet contains 250 mg of gefitinib.
Gefitinib is a white powder and a free base. It has pKas of 5.4 and 7.2 and therefore ionises progressively in solution as the pH falls. Gefitinib can be defined as sparingly soluble at pH 1, but is practically insoluble above pH 7, with the solubility dropping sharply between pH 4 and 6.
Gefitinib is achiral.
Excipients with known effect: Each tablet contains lactose monohydrate. For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Film-coated tablets (tablet).
The tablets are brown, round, biconvex, impressed with "Iressa 250" on one side and plain on the other.
4 Clinical Particulars
4.1 Therapeutic Indications
Treatment of patients with locally advanced or metastatic Non Small Cell Lung Cancer (NSCLC) whose tumours express activating mutations of the EGFR tyrosine kinase.
4.2 Dose and Method of Administration
The recommended dose of Iressa is one 250 mg tablet once a day, taken with or without food. If a dose of Iressa is missed, it should be taken as soon as the patient remembers. If it is less than 12 hours to the next dose, the patient should not take the missed dose. Patients should not take a double dose (two doses at the same time) to make up for a forgotten dose.
Where dosing of whole tablets is not possible, such as patients who are only able to swallow liquids, tablets may be administered as a dispersion in water. The tablet should be dropped into half a glass of drinking water (non-carbonated), without crushing, and the glass stirred until the tablet has dispersed (approximately 15 minutes) and the contents subsequently drunk immediately. The glass should be rinsed with a further half glass of water and the contents drunk. The liquid can also be administered via a nasogastric tube.
Dosage adjustment. No dosage adjustment is required on the basis of patient age, body weight, gender, ethnicity, renal function or in patients with moderate to severe hepatic impairment due to liver metastases. Gefitinib exposure is increased in patients with moderate to severe hepatic impairment due to cirrhosis or hepatitis (see Section 5.2 Pharmacokinetic Properties). These patients should be closely monitored for adverse events.
Patients with poorly tolerated diarrhoea or skin adverse drug reactions may be successfully managed by providing a brief (up to 14 days) therapy interruption followed by reinstatement of the 250 mg dose.
4.3 Contraindications
Known severe hypersensitivity to the active substance or to any of the excipients of this product.
4.4 Special Warnings and Precautions for Use
EGFR mutation assessment. When considering the use of Iressa as first line treatment for advanced or metastatic NSCLC, it is recommended that EGFR mutation assessment of the tumour tissue is attempted for all patients. When assessing the mutation status of a patient it is important that a well validated and robust methodology is chosen to minimise the possibility of false negative or false positive determinations. Tumour samples which are used for the diagnosis of advanced NSCLC are the preferred sample type for EGFR mutation testing. A tumour sample should be collected and tested where possible. If a tumour sample is not available or evaluable, then circulating tumour DNA (ctDNA) obtained from a blood (plasma) sample may be used. Only robust, reliable, sensitive test(s) with demonstrated utility on ctDNA should be used for the determination of EGFR mutation status of ctDNA. Analytical performance characteristics with respect to tissue results (specificity, sensitivity, negative predictive value and positive predictive value) should be equivalent or better than that obtained with the Qiagen Therascreen EGFR RGQ PCR kit used in the IFUM study (see Section 5.1 Pharmacodynamic Properties). EGFR mutations identified in ctDNA are highly predictive of EGFR mutation positive tumours. However it is not always possible to detect EGFR mutations using this sample type (0.2% false positives, 34.3% false negatives) (see Section 5.1 Pharmacodynamic Properties).
Interstitial lung disease. Interstitial Lung Disease (ILD), which may be acute in onset, has been observed in patients receiving Iressa, and some cases have been fatal. Patients typically have an acute onset of dyspnoea associated with cough, low grade fever, respiratory distress and arterial oxygen desaturation. Symptoms may become severe within a short time. If patients present with worsening of respiratory symptoms such as dyspnoea, cough and fever, Iressa should be interrupted and prompt investigation initiated. Radiological investigations frequently show pulmonary infiltrates or interstitial shadowing with ground glass appearance. If ILD is confirmed, Iressa should be discontinued and the patient treated appropriately.
In the placebo-controlled ISEL trial (1692 patients), the incidence of ILD-type events in the overall population was similar and approximately 1% in both treatment arms. The majority of ILD type event reports were from patients of Oriental ethnicity and the ILD incidence among patients of Oriental ethnicity receiving Iressa therapy and placebo was similar approximately 3 and 4% respectively. One ILD-type was fatal, and this occurred in a patient receiving placebo.
In the INTEREST trial (1466 patients), the incidence of ILD was 1.4% in the Iressa group and 1.1% in the docetaxel group. One ILD event was fatal and this occurred in a patient receiving Iressa.
In the IPASS trial (1217 patients) in Asian patients, the incidence of ILD-type events was 2.6% on the Iressa treatment arm versus 1.4% on the carboplatin/ paclitaxel treatment arm.
In a Japanese pharmacoepidemiological case control study (see Section 4.8 Adverse Effects (Undesirable Effects)) in 3159 patients with NSCLC who were followed up for 12 weeks when receiving Iressa or chemotherapy, the following risk factors for developing ILD (irrespective of whether the patient received Iressa or chemotherapy) were identified: smoking, poor performance status (PS ≥ 2), CT scan evidence of reduced normal lung (≤ 50%), recent diagnosis of NSCLC (< 6 months), pre-existing ILD, increasing age (> 55 years old) and concurrent cardiac disease. Risk of mortality among patients who developed ILD on both treatments was higher in patients with the following risk factors: smoking, CT scan evidence of reduced normal lung (≤ 50%), pre-existing ILD, increasing age (≥ 65 years old), and extensive areas adherent to pleura (≥ 50%).
Cerebrovascular events. Cerebrovascular events have been reported in clinical studies of Iressa. A relationship with Iressa has not been established. An increased risk of cerebral haemorrhage in adult patients with NSCLC receiving Iressa has not been established.
In a phase I/II trial of Iressa and radiation in paediatric patients, newly diagnosed with brain stem glioma or incompletely resected supratentorial malignant glioma, 4 cases (1 fatal) of CNS haemorrhages were reported from the 45 patients enrolled. A further case of CNS haemorrhage in a child with an ependymoma from a trial with Iressa alone has been reported.
Severe or persistent diarrhoea, nausea, vomiting or anorexia. Patients should be advised to seek medical advice promptly in the event of developing severe or persistent diarrhoea, nausea, vomiting or anorexia (see Section 4.8 Adverse Effects (Undesirable Effects)). These symptoms should be managed as clinically indicated.
Eye symptoms. Patients presenting with signs and symptoms suggestive of keratitis such as acute or worsening: eye inflammation, lacrimation, light sensitivity, blurred vision, eye pain and/or red eye should be referred promptly to an ophthalmology specialist.
If a diagnosis of ulcerative keratitis is confirmed, treatment with Iressa should be interrupted, and if symptoms do not resolve, or recur on reintroduction of Iressa, permanent discontinuation should be considered.
Combination cytotoxic therapy. Randomised controlled trials have demonstrated that Iressa combined with doublet, platinum based cytotoxic chemotherapy in advanced NSCLC provides no added benefit over the cytotoxic chemotherapy alone. Iressa should therefore only be used as monotherapy in advanced NSCLC in patients who have previously received treatment with cytotoxic chemotherapy.
Gastrointestinal perforation. Gastrointestinal perforation has been reported in patients taking Iressa. In most cases this is associated with other known risk factors including increasing age, concomitant medications such as steroids or NSAIDs, underlying history of GI ulceration, smoking or bowel metastases at sites of perforation.
QT interval prolongation. Data from non-clinical (in vitro) studies indicate that gefitinib has the potential to inhibit the cardiac action potential repolarization process (e.g. QT interval). However, safety data obtained from clinical trials and post-marketing surveillance have not suggested any adverse cardiac effects of gefitinib.
Severe cutaneous reactions. Severe cutaneous reactions have been reported with gefitinib including erythema multiforme and life-threatening reactions such as Stevens-Johnson syndrome or toxic epidermal necrolysis (e.g. progressive skin rash often with blisters or mucosal lesions). Treatment with gefitinib should be interrupted if the patient develops severe bullous, blistering or exfoliative conditions, and discontinued if Stevens-Johnson syndrome or toxic epidermal necrolysis are confirmed.
Use in hepatic impairment. Liver function test abnormalities (including increases in alanine aminotransferase, aspartate aminotransferase, bilirubin) have been observed (see Section 4.8 Adverse Effects (Undesirable Effects)) uncommonly presenting as hepatitis. There have been isolated reports of hepatic failure which in some cases led to fatal outcomes. Therefore, periodic liver function testing is recommended. Iressa should be used cautiously in the presence of mild to moderate changes in liver function. Discontinuation should be considered if changes are severe.
Use in renal impairment. There have been reports of renal failure secondary to dehydration due to diarrhoea, nausea, vomiting and/or anorexia, or associated with pre-renal factors such as concurrent infections or concomitant medications including chemotherapy. In more severe or persistent cases of diarrhoea, or cases leading to dehydration, particularly in patients with known risk factors (e.g. renal disease, concurrent vomiting, concomitant medications that impair ability to tolerate dehydration such as NSAIDs and diuretics), Iressa therapy should be interrupted and appropriate measures taken to intensively rehydrate the patient. In addition, urea, electrolytes and creatinine should be monitored in patients at high risk of dehydration.
Paediatric use. Iressa is not recommended for use in children or adolescents as safety and effectiveness in these patient populations has not been studied.
Use in the elderly. No data available.
Effects on laboratory tests. Liver function test abnormalities (including increases in alanine aminotransferase, aspartate aminotransferase, bilirubin) have been observed (see Section 4.8 Adverse Effects (Undesirable Effects)) uncommonly presenting as hepatitis. There have been isolated reports of hepatic failure which in some cases led to fatal outcomes.
4.5 Interactions with Other Medicines and Other Forms of Interactions
In vitro studies with human hepatic microsomes have shown that the metabolism of Iressa is mainly via the CYP3A4 isoform of the hepatic cytochrome P-450 system. Iressa may be expected to interact with other drugs that induce, inhibit or are metabolised by this system. Iressa showed little enzyme induction effect in animal studies and in vitro studies have shown that Iressa has limited potential to inhibit CYP2D6.
The clinically or potentially significant clinical drug interactions between Iressa and the following drugs/ drug classes are described below.
Other drugs that affect Iressa. Demonstrated interactions. Medicines that inhibit CYP3A4. Co-administration with itraconazole (a CYP3A4 inhibitor) resulted in an 80% increase in the mean AUC of Iressa in healthy volunteers. This increase may be clinically relevant since adverse experiences are related to dose and exposure. Although interaction studies with other CYP3A4 inhibitors have not been performed it is expected that drugs such as ketoconazole, clotrimazole, ritonavir would also inhibit Iressa metabolism.
Medicines (e.g. ranitidine) that increase gastric pH. In a trial in healthy volunteers co-administration of ranitidine at a dose that caused sustained elevations in gastric pH, ≥ 5, resulted in a reduced mean Iressa AUC by 47%. As a consequence, drugs that cause significant sustained elevations in gastric pH may reduce plasma concentrations of Iressa and therefore may reduce efficacy.
Rifampicin. Co-administration with rifampicin (a known potent CYP3A4 inducer) in healthy volunteers reduced mean Iressa AUC by 83% of that without rifampicin.
Theoretical interactions. Other CYP3A4 inducers. Substances that are inducers of CYP3A4 activity may increase metabolism and decrease Iressa plasma concentrations. Therefore, co-medication with CYP3A4 inducers (e.g. phenytoin, carbamazepine, barbiturates or St John's wort) may reduce efficacy.
Effects of Iressa on other medicines. Demonstrated interactions. Medicines metabolised by CYP2D6. In a clinical trial in patients, Iressa was co-administered with metoprolol (a CYP2D6 substrate). This resulted in a mean 35% increase in exposure to metoprolol. Iressa may increase the blood concentrations of other co-administered drugs metabolised by CYP2D6.
Theoretical interactions. Warfarin. Although no formal drug interaction study has been conducted, elevations in International Normalised Ratio (INR) and/or bleeding events have been reported in some patients taking warfarin. Consistent with standard anticoagulant therapy practice, patients taking warfarin should be monitored regularly for changes in prothrombin time or INR (see Section 4.8 Adverse Effects (Undesirable Effects)).
Vinorelbine. Phase II clinical trial data, where Iressa and vinorelbine have been used concomitantly, indicate that Iressa may exacerbate the neutropenic effect of vinorelbine.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility. Gefitinib given at 20 mg/kg/day (0.7-fold the clinical dose based on body surface area) for 4 weeks prior to mating until day 7 of gestation affected ovulation in female rats, resulting in a reduction in the number of corpora lutea.
Use in pregnancy. (Category C)
There are no data from the use of Iressa in pregnant women. When gefitinib was administered during organogenesis, an increase in the incidence of incomplete ossifications was observed in rats and reduced foetal weights were observed in rabbits at maternally toxic doses. Malformations were not observed in rats; they were observed in rabbits only at a severely maternally toxic dose. Women of childbearing potential must be advised to avoid becoming pregnant while receiving Iressa therapy.
Use in lactation. Mothers must be recommended to discontinue breast-feeding while receiving Iressa therapy.
There are no data from the use of Iressa in breast-feeding women. It is not known whether gefitinib or its metabolites are excreted in human milk. However, in lactating rats administered 5 mg/kg orally (about 0.2-fold the clinical dose based on body surface area), gefitinib and some metabolites were excreted in milk. When gefitinib was administered to the rat during gestation and lactation, there was a reduction in pup survival at a dose of 20 mg/kg/day (about 0.7-fold the clinical dose based on body surface area).
4.7 Effects on Ability to Drive and Use Machines
During treatment with Iressa, asthenia has been reported and those patients who experience this symptom should observe caution when driving or using machines.
4.8 Adverse Effects (Undesirable Effects)
Reporting suspected adverse effects. Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
The most commonly reported adverse drug reactions (ADRs), occurring in more than 20% of the patients receiving Iressa 250 mg are diarrhoea, rash, pruritus, dry skin and acne. ADRs usually occur within the first month of therapy and are generally reversible. Approximately 10% of patients had a severe ADR (Common Toxicity Criteria, (CTC) grade 3 or 4). However, approximately 3% of patients stopped therapy due to an ADR.
In a phase II trial, Japanese patients experienced a higher frequency of ADRs compared to non-Japanese patients. Because of possible confounding factors in the two patient groups, it is uncertain if this difference is due to ethnic factors.
ADRs have been assigned to the frequency categories where possible based on the incidence of comparable Adverse Event reports in a pooled dataset from the IPASS, ISEL and INTEREST phase III clinical trials (2462 Iressa-treated patients). In assigning these frequencies no account was taken of the frequency of reports within the comparative treatment groups or whether the investigator considered it to be related to study medication.
Frequencies of occurrence of undesirable effects are defined as: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000), not known (cannot be estimated from the available data). Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness. See Table 1.

Severe cutaneous reactions have been reported with gefitinib including erythema multiforme and life-threatening reactions such as Stevens-Johnson syndrome or toxic epidermal necrolysis (e.g. progressive skin rash often with blisters or mucosal lesions).
4.9 Overdose
For information on the management of overdose, contact the Poison Information Centre on 131126 (Australia).
There is no specific treatment in the event of overdose of Iressa. In phase I clinical trials, a limited number of patients were treated with daily doses of up to 1,000 mg. An increase of frequency and severity of some adverse reactions was observed, mainly diarrhoea and skin rash. Adverse reactions associated with overdose should be treated symptomatically; in particular severe diarrhoea should be managed as clinically indicated.
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action. Gefitinib is a selective inhibitor of the epidermal growth factor receptor (EGFR) tyrosine kinase, commonly expressed in solid human tumours of epithelial origin. Gefitinib has been demonstrated to inhibit the growth of a wide range of human tumour xenografts in nude mice, to inhibit angiogenesis in xenografts, and to increase apoptosis and inhibit invasiveness and secretion of angiogenic factors in human cancer cell lines in vitro. In animal or in vitro studies, gefitinib has also been demonstrated to enhance the anti-tumour activity of chemotherapy, radiotherapy and hormonal therapy.
Resistance. Most NSCLC tumours with sensitizing EGFR kinase mutations eventually develop resistance to gefitinib treatment with a median time to disease progression of around 1 year. In about 60% of cases, resistance is associated with a secondary T790M mutation for which T790M targeted EGFR TKIs may be considered as a next line treatment option. Other potential mechanisms of resistance that have been reported following treatment with EGFR signal blocking agents include bypass signaling such as HER2 and MET gene amplification and PIK3CA mutations. Phenotypic switch to small cell lung cancer has also been reported in 5-10% of cases.
Clinical trials. Phase III studies. IPASS. In a phase III clinical trial conducted in Asia in 1217 patients with advanced (stage IIIB or IV) NSCLC of adenocarcinoma histology who were ex-light (ceased smoking > 15 years ago and smoked < 10 pack years) or never smokers and had not received previous chemotherapy, Iressa was proven to be superior to carboplatin (AUC 5.0 or 6.0)/ paclitaxel (200 mg/m2) in terms of Progression Free Survival (PFS) (Hazard Ratio [HR] 0.74, 95% CI 0.65 to 0.85, p < 0.0001), the primary endpoint of the trial. Patients were randomised to either Iressa 250 mg once daily or carboplatin/ paclitaxel AUC 5 or 6 mg/mL/min/200 mg/m2 iv every 3 weeks. The effect was not constant over time, initially favouring carboplatin/ paclitaxel and then favouring gefitinib, potentially driven by differences in PFS outcomes by EGFR mutation status. EGFR mutation status was a strong predictive biomarker for the effect of gefitinib compared to carboplatin/ paclitaxel. In subgroup analysis of PFS by EGFR mutation status, gefitinib was significantly better than carboplatin/ paclitaxel in patients with mutation positive tumours whereas carboplatin/ paclitaxel was significantly better than gefitinib in patients with mutation-negative tumours (see Table 2).
An analysis of overall survival (OS) was performed after 954 deaths (78% maturity), which demonstrated no statistically significant difference in OS for Iressa versus carboplatin/ paclitaxel in the overall study population (see Table 2). The analysis was confounded by treatment on progression. Approximately half the patients progressing on gefitinib were transferred to carboplatin/ paclitaxel and approximately half the patients progressing on carboplatin/ paclitaxel were transferred to an EGFR tyrosine kinase inhibitor.



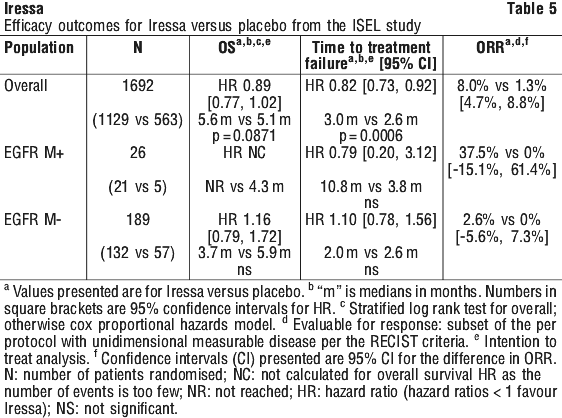
ISEL. The ISEL study was a phase III randomised, double blind, parallel group clinical trial comparing Iressa 250 mg once daily plus BSC versus placebo plus BSC in 1692 pre-treated patients with locally advanced or metastatic NSCLC who had received 1 or 2 prior chemotherapy regimens and were refractory (90%) or intolerant to their most recent regimen (see Table 5).
IFUM study. The IFUM study was a single arm, multicentre study conducted in Caucasian patients (n = 106) with activating, sensitizing EGFR mutation positive NSCLC to confirm that the activity of gefitinib is similar in Caucasian and Asian populations. The ORR according to investigator review was 70% and the median PFS was 9.7 months. These data are similar to those reported in the IPASS study.
Circulating tumour DNA (ctDNA). In the IFUM trial, mutation status was assessed in tumour and ctDNA samples derived from plasma, using the Therascreen EGFR RGQ PCR kit (Qiagen) (see Section 4.4 Special Warnings and Precautions for Use). Both ctDNA and tumour samples were evaluable for 652 patients out of 1060 screened. The sensitivity of EGFR mutation testing in ctDNA using the Qiagen Therascreen EGFR RGQ PCR kit was 65.7%, with a specificity of 99.8% relative to the matched tissue results using the same Qiagen Therascreen kit. The positive and negative predictive values of ctDNA were 98.6% and 93.8%, respectively (see Table 6). Objective response rate in the IFUM full analysis set population was 69.8% (95% CI: 60.5% to 77.7%). The ORR in those patients within the full analysis set population who were ctDNA mutation positive was 77.3% (95% CI: 65.8 to 85.7).

5.2 Pharmacokinetic Properties
Following intravenous administration, gefitinib is rapidly cleared, extensively distributed and has a mean elimination half-life of 48 hours. Following oral dosing in cancer patients, absorption is moderately slow and the mean terminal half-life is 41 hours. Administration of gefitinib once daily results in a 2 to 8-fold accumulation with steady state exposure achieved after 7 to 10 doses. At steady state, the ratios of Cmax to Cmin are typically maintained within a 2 to 3-fold range over the 24-hour dosing interval.
Absorption. Following oral administration of Iressa, peak plasma concentrations of gefitinib typically occur at 3 to 7 hours after dosing. Mean absolute bioavailability is 59% in cancer patients. Exposure to gefitinib is not significantly altered by food.
Distribution. Mean volume of distribution at steady state of gefitinib is 1,400 L indicating extensive distribution into tissue. Plasma protein binding is approximately 90%. Gefitinib binds to serum albumin and α1-acid glycoprotein.
Metabolism. In vitro data indicate that CYP3A4 is the major P450 isozyme involved in the oxidative metabolism of gefitinib. In vitro studies have shown that gefitinib has limited potential to inhibit CYP2D6 (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Three sites of biotransformation have been identified in the metabolism of gefitinib: metabolism of the N-propylmorpholino-group, demethylation of the methoxy-substituent on the quinazoline and oxidative defluorination of the halogenated phenyl group.
The major metabolite identified in human plasma is O-desmethyl gefitinib. It was 14-fold less potent than gefitinib at inhibiting EGFR stimulated cell growth and it is therefore unlikely that it contributes significantly to the clinical activity of gefitinib.
The production of O-desmethyl gefitinib has been shown, in vitro, to be via CYP2D6. The role of CYP2D6 in the metabolic clearance of gefitinib has been evaluated in a clinical trial in healthy volunteers genotyped for CYP2D6 status. In poor metabolisers, no measurable levels of O-desmethyl gefitinib were produced. The range of gefitinib exposures achieved in both the extensive and the poor metaboliser groups were wide and overlapping but the mean exposure to gefitinib was 2-fold higher in the poor metaboliser group. The higher average exposures that could be achieved by individuals with no active CYP2D6 may be clinically relevant since adverse experiences are related to dose and exposure.
Excretion. Gefitinib total plasma clearance is approximately 500 mL/min. Excretion is predominantly via the faeces with renal elimination of drug and metabolites accounting for less than 4% of the administered dose.
Special populations. Population kinetics. In population based data analyses in cancer patients, no relationships were identified between predicted steady state trough concentration and patient age, body weight, gender, ethnicity or creatinine clearance.
Hepatic insufficiency. In a phase I open-label study of single dose gefitinib 250 mg in patients with mild, moderate or severe hepatic impairment due to cirrhosis (according to Child-Pugh classification), there was an increase in exposure in all groups compared with healthy controls. An average 3.1-fold increase in exposure to gefitinib in patients with moderate and severe hepatic impairment was observed. None of the patients had cancer, all had cirrhosis and some had hepatitis. This increase in exposure may be of clinical relevance since adverse experiences are related to dose and exposure to gefitinib.
Gefitinib has been evaluated in a pharmacokinetic study in 31 patients with solid tumours and normal (14 patients), moderate (13 patients) or severe (4 patients) hepatic dysfunction due to liver metastases. It was shown that following daily dosing of 250 mg Iressa for 28 days, time to steady state, total plasma clearance and steady state exposure (Cmaxss, AUC24ss) were similar for the groups with normal and moderately impaired hepatic function. Data from the 4 patients with severe hepatic dysfunction due to liver metastases suggested that steady-state exposures in these patients are also similar to those with normal hepatic function.
Other populations. Patients that have never smoked, have adenocarcinoma histology, are female gender or are of Asian ethnicity, are associated with a higher rate of EGFR mutation positive tumours.
5.3 Preclinical Safety Data
Genotoxicity. There was no evidence for a genotoxic potential of gefitinib in assays for gene mutation (bacteria and mammalian cells in vitro) and clastogenicity (mammalian cells in vitro and in vivo rat micronucleus test).
Carcinogenicity. A 2-year oral (gavage) carcinogenicity study in rats resulted in a small but statistically significant increase in the incidence of hepatocellular adenomas in both male and female rats and mesenteric lymph node haemangiosarcomas in female rats at the high dose of 10 mg/kg/day. An increased incidence of hepatocellular adenomas was also seen in a 2-year oral (gavage) carcinogenicity study in mice dosed at 50 mg/kg/day and above. The dose of 10 mg/kg/day in rats and 50 mg/kg/day in mice was associated with systemic exposure (based on plasma AUC) of approximately two (2) and three (3) times that anticipated in patients at the recommended clinical dose. The non-effect-dose-level (NOEL) for these effects was 5 mg/kg/day in rats and 10 mg/kg/day in mice, which was associated with systemic exposure of approximately 1.2 and 0.3 times the anticipated value in patients at the recommended clinical dose.
6 Pharmaceutical Particulars
6.1 List of Excipients
The tablets contain the following excipients: lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, povidone, sodium lauryl sulfate, magnesium stearate, hypromellose, macrogol 300, titanium dioxide, iron oxide yellow CI77492 and iron oxide red CI77491.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C.
6.5 Nature and Contents of Container
Iressa is available in packs of 30 tablets in PVC/aluminium blisters.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
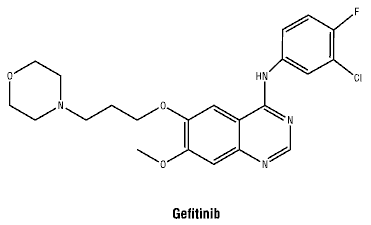
Molecular formula: C22H24ClFN4O3.
Molecular weight: 446.90.
CAS number. 184475-35-2.
7 Medicine Schedule (Poisons Standard)
Prescription only medicine (Schedule 4).
Date of First Approval
28 April 2003
Date of Revision
13 January 2021
Summary Table of Changes

Reasonable care is taken to provide accurate information at the time of creation. This information is not intended as a substitute for medical advice and should not be exclusively relied on to manage or diagnose a medical condition. The Australian Commission on Safety and Quality in Health Care disclaims all liability (including for negligence) for any loss, damage, injury or any other negative effects resulting from reliance on or use of this information. Read our full disclaimer. This website uses cookies. Read our privacy policy.