Australian Unique Device Identification (UDI) system
Australia has established a Unique Device Identification (UDI) system for medical devices supplied in Australia.
Overview
The Australian Government is strengthening patient safety by introducing Unique Device Identification (UDI) for medical devices and in vitro diagnostic medical devices.
UDI allows clear and unambiguous identification of medical devices and facilitates access to additional information about the device. It will provide a range of benefits to healthcare organisations, healthcare professionals and patients – strengthening patient safety and supporting quicker identification of and responses to device safety issues.
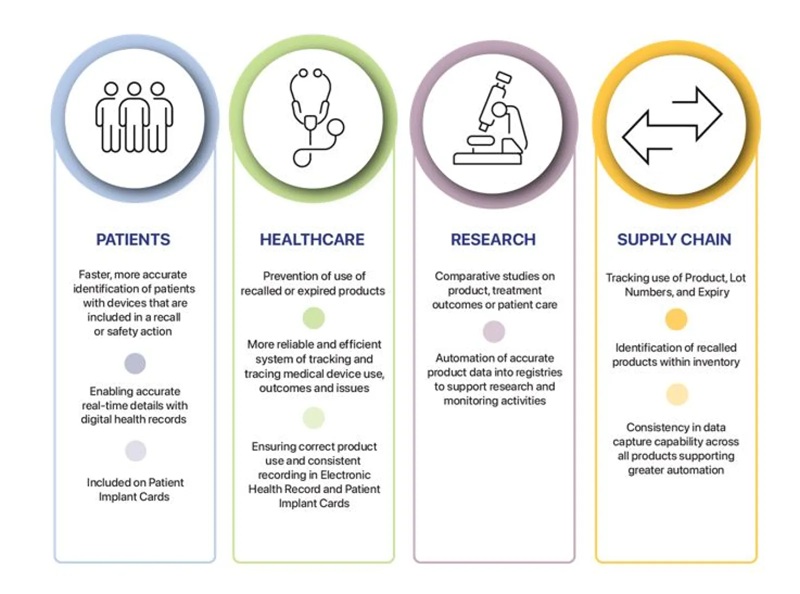
UDI will support healthcare by making it easier to identify, manage and monitor medical devices within clinical workflows and information systems and assist in recording and tracking specific devices used on or implanted into patients. UDI supports the identification and removal of problematic devices from storage and distribution and prevents further use.
Medical device manufacturers and sponsors must start applying the UDI to the labels of high‑risk medical devices from July 2026, followed by other classes of medical devices and in vitro diagnostic devices in later years. They must also provide the UDI-Device Identifier and additional information on the devices into a centralised database managed by the Therapeutic Goods Administration (TGA), known as the Australian UDI Database (AusUDID). This database can be accessed by healthcare professionals and patients.
The Commission is collaborating with the TGA to integrate the UDI across all stages of the healthcare system by capturing the UDI in processes and systems that manage product or patient data. The Commission worked with the TGA, the Australian Government Department of Health, Disability and Ageing and Queensland Health in developing and piloting the use of UDI.
Learn more
For more information on the benefits of implementing UDI and how it can be integrated into processes that span the device lifecycle, see UDI for Australian healthcare: Executive summary (TGA website).
The TGA has also published a range of resources on their UDI Hub, including: